
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

The exome is commonly defined as encompassing all exons within protein-coding genes, inclusive of elements that are not protein-encoding, such as sequences of microRNA or lncRNA. Examining the exome aids in the identification of loci that may precipitate specific diseases. For the investigation of rare Mendelian disorders, exome sequencing proves to be a particularly proficient method for the detection of genetic variations.
Accelerated by advancements in targeted enrichment strategies and breakthroughs in DNA sequencing technologies, the evolution of whole exome sequencing (WES) has been instrumental. WES represents a genomic analysis technique geared towards the detailed investigation of all transcribable exons within a genome. This is accomplished by leveraging DNA microarrays or whole-genome sequencing platforms in tandem with sequence capture technologies. These tools allow for the selective capture and enrichment of exon regions in the DNA, which are then subject to high-throughput sequencing for insight into gene expression regulatory mechanisms.
WES exhibits the capacity to detect potential mutations that may be associated with phenotypes or diseases, as well as overall genomic variations, novel gene discovery, and the phenotype-to-genotype relationships of gene mutations.
WES entails the extraction of all exonic regions, the prime dominions orchestrating protein synthesis. Proteins form the structural and functional core of the human body, rendering the exome zones within DNA of greatest genetic value. Given that exons constitute merely 1% of an individual's genome, WES successfully manages to control costs while amplifying testing depth—a critical factor bearing on accuracy. Significant academic research outputs are currently centered on the exonic region, thus data generated from WES are adequately substantial for future updates. With the advent of fresh research findings, direct data analysis can be employed without necessitating a re-test.
Whole Genome Sequencing (WGS) endeavors to capture all genetic loci. Areas beyond exons, which account for 99% of total loci, contain abundant repetitive and nonsensical segments. The yield of relevant scholarly literature is also markedly skimpy, implying that the legible content derived from WGS is nearly indistinguishable from WES-extracted content. However, significant costs are disproportionately squandered on 'pointless' loci in WGS, making it challenging to guarantee sequencing depth. In certain WGS genetic tests that cost several thousand, the average depth is a mere 30X. For the loci under analysis, the sequencing depth could dwindle to single-figure levels, rendering the data practically valueless for reference.
Compared with WGS, WES commands an advantage in the following three facets:
Therefore, WES is currently considered the most cost-effective gene testing technique.
Services you may interested in
Whole Exome Sequencing as a genomic analytical technique seeks genetic mutations tied to protein function variations. The basic principle encompasses:
DNA Capture and Enrichment: DNA or RNA probes specific to exon regions are first used to capture and enrich DNA sequences. Typically this is achieved through liquid-phase hybrid capture technology, which employs the principles of base pairing to hybridize biotin-labelled RNA probes with DNA libraries with adapter sequences, and then enriches the DNA of the target area through magnetic bead binding.
High-Throughput Sequencing: The enriched DNA sequences are then sequenced via high-throughput sequencing technology. Sequencing is the process of discovering all deoxyribonucleotide arrangements in the exome, which may help us understand potential pathophysiological changes in certain diseases.
Data Analysis: The sequencing results are subjected to bioinformatic analysis to pinpoint genetic mutations associated with protein function variations.
The conventional working process of Whole Exome Sequencing (WES) includes sample preparation, exome capture and library design, target enrichment, sequencing, and bioinformatic analysis. An illustration catering to the WES workflow can be depicted below.
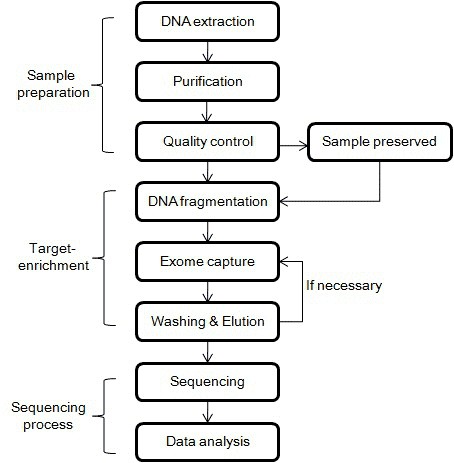 Figure1. Workflow of WGS.
Figure1. Workflow of WGS.
Notice:The precise protocols differ across various types of samples, reagent kits, and sequencing equipment. Researchers ought to adhere to the instructions specified for the reagents, kits, and sequencing machinery in use.
Sample preparation: whole blood samples, peripheral blood mononuclear cells (PBMCs), freshly frozen tissues, formalin-fixed paraffin-embedded (FFPE) samples, plasma samples, and amniotic fluid cells, among others, can be used for exome sequencing. DNA is extracted from the biological samples to be tested and fragmented through physical methods or enzymatic processes. Physical disruption includes shearing, sonication, and fluid dynamic shearing, where shearing and sonication serve as the primary methods of DNA fragmentation. Enzymatic processes typically involve employing nucleases or transposases, which fragment the DNA into smaller pieces. Nucleases induce the breakage of phosphodiester bonds within nucleic acids, resulting in DNA fragmentation.
Capture and enrichment of exons: During exome capture and enrichment, exons are selectively enriched using exome capture technology. The fundamental concept behind target enrichment pertains to exploiting the physicochemical differences between the target compound and other substances to facilitate their separation. Following the isolation of the exome from the rest of the genome, multiple washing stages are required to obtain a purer exome. Although distilled water is typically used to elute the targets, certain specialized kits may necessitate specific elution solutions.
Library construction: Library construction involves repairing the ends of the enriched exonic DNA fragments, attaching connectors, and amplifying the libraries to convert the enriched exonic DNA fragments into sequencing libraries—a step that provides ample DNA templates for subsequent sequencing.
Table1. Common kits of target-enrichment for sequencing.
| Kits | Targeted Region | Genomic DNA Input Required | Adapter Addition | Probe Length (mer) |
| Agilent SureSelect XT2 V6 Exome | 60 Mb | 100 ng | Ligation | 120 |
| Agilent SureSelect XT2 V5 Exome | 51 Mb | 100 ng | Ligation | 120 |
| IDT xGEN Exome Panel | 39 Mb | 500 ng | Ligation | not described |
| Illumina Nextera Rapid Capture Expanded Exome | 62 Mb | 50 ng | Transposase | 95 |
| Roche NimblegenSeqCap EZ Exome v3.0 | 64 Mb | 1 ug | Ligation | 60 - 90 |
Sequencing: With advancements in sequencing techniques, Next-Generation Sequencing (NGS) is more extensively employed for exome sequencing. The rationale behind NGS involves coupling the exome sample to an appropriate base (such as flowcell in Illumina Hiseq and magnetic beads in Roche-454), followed by in situ PCR duplication to ensure signal amplification in every round. The ddNTP is examined after each round of extension, and the complete sequence is eventually compiled using a bioinformatics algorithm. The high efficiency and throughput capabilities render NGS an appealing option for high-throughput sequencing.
Apart from NGS, third-generation sequencing technologies are evolving rapidly and possess efficiencies that vastly outshine NGS. Single-molecule sequencing, a distinctive feature of third-generation sequencing, can dramatically trim the time and cost associated with whole-genome sequencing to mere minutes. Companies such as PacificBio and Oxford Nanopore have demonstrated the efficiency of their methods, which could potentially incite a revolution in the realm of exome sequencing.
Table2. Common methods used for sequencing nowadays.
| Methods | Company | Generation | Read length | Accuracy | Reads per run | Time per run |
| Ion semiconductor | Ion Torrent | 2nd generation | Up to 600 bp | 99.60% | up to 80 million | 2 hours |
| Pyrosequencing(454) | Roche | 2nd generation | 700 bp | 99.90% | 1 million | 24 hours |
| Sequencing by synthesis | Illumina | 2nd generation | 75-300 bp | 99.90% | 1 million to 3 billion | 1 to 11 days |
| Sequencing by ligation (SOLiD) | ABI | 2nd generation | 50+35 or 50+50 bp | 99.90% | 1.2 to 1.4 billion | 1 to 2 weeks |
| Nanopore Sequencing | Oxford Nanopore Technologies | 3rd generation | up to 500 kb | 92–97% (single read)* | dependent on read length selected by user | 1 min to 48 hours |
| Single-molecule real-time sequencing | Pacific Biosciences | 3rd generation | 30,000 bp | 87% (single read)* | 10-20 billion | 0.5-20 hours |
Data analysis: The data analysis process involves eliminating low-quality reads during sequencing through quality control, aligning high-quality reads with the reference genome, quantifying single nucleotide polymorphisms (SNPs), insertions and deletions (Indels), and copy number variations (CNVs), followed by annotation, screening, analysis, and validation. Data analysis constitutes a critical juncture in exome sequencing, encompassing the evaluation and processing of sequencing data quality, aligning sequencing reads to the reference genome, and identifying genetic mutations within the samples. The detected genetic mutations need to be annotated, including discerning the function of the mutation, its pathological significance, and its potential impact.
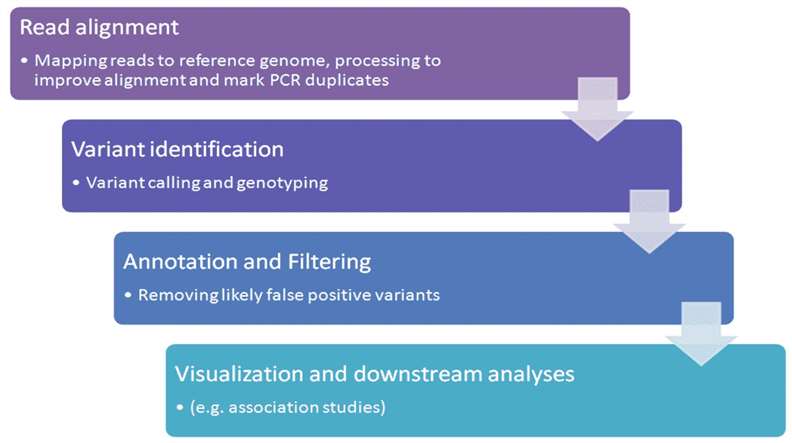 Figure2. The typical variant calling pipeline.
Figure2. The typical variant calling pipeline.
Cost-effectiveness: Compared to Whole Genome Sequencing, Whole Exome Sequencing offers deeper coverage, greater accuracy, and is more economically efficient.
High sequencing depth: The sequencing depth can reach more than 120x.
High throughput: It meets the research needs of multiple target regions from a large number of samples.
High accuracy: With deep sequencing coverage, data accuracy is high and efficient.
Whole exome sequencing (WES) finds applications in various domains of biomedical research, including the study of monogenic inherited diseases, complex diseases, somatic mutations in tumors, discovery of oncogenes and tumor suppressor genes, and advancement of personalized medicine initiatives.
Tumors, being complex diseases instigated by multiple gene mutations, can be effectively understood with the help of whole exome sequencing. It enables scientists and clinicians to uncover variations in tumor-related genes to achieve accurate diagnosis and prognosis of the tumor. This primarily includes research into driver genes, molecular sub-typing of tumors, recurrent metastasis research, and tailored medication for individual tumors.
Genetic disorders frequently originate from gene mutations, often occurring in the exonic regions that encode proteins. Traditional mutation detection methods require a thorough investigation of disease-related genes one at a time, which is time-consuming, labor-intensive, and necessitates a substantial sample size. However, exome sequencing mitigates these drawbacks by simultaneously investigating multiple disease genes, resulting in significantly improved detection efficiency and accuracy. By applying WES, we can sequence all exonic regions required to encode proteins, subsequently identifying gene mutations or polymorphic loci related to the disease onset. This facilitates accurate diagnosis and treatment of genetic disorders.
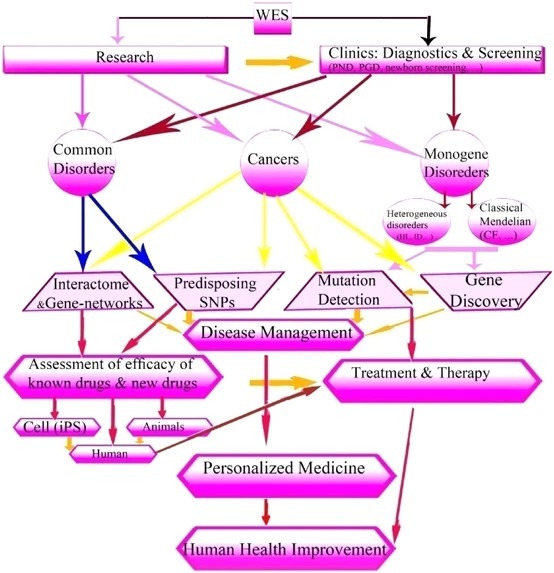 Figure3. WES and impact of its genetic consequences on human public health. (Rabbani B et al., 2014)
Figure3. WES and impact of its genetic consequences on human public health. (Rabbani B et al., 2014)
WES holds significant intrigue for clinical applications, primarily because it encompasses the actionable regions within a genome. The primary objective is to identify variations within the exonic regions and determine the causal mutations for diseases or pathogenic variants. The intersection of big data analysis and WES is driving advances in personalized medicine in numerous dimensions.
If you are interested in our genomics services, please feel free to contact our scientists. We are more than happy to be of assistance. In addition to genomics sequencing, we also provide services including transcriptomics, epigenomics, microbial genomics, single-cell sequencing, and PacBio SMRT sequencing.
References:


