
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Gene Set Enrichment Analysis (GSEA) serves as an advanced computational tool frequently employed for the analysis of genomic data and transcriptomic data. This method determines if particular collections of genes, termed gene sets, exhibit statistically meaningful variations in expression levels when comparing two distinct biological states. By concentrating on the collective behavior of genes rather than analyzing them in isolation, GSEA facilitates a deeper understanding of the overarching biological processes. Consequently, researchers are better equipped to decipher the implications of extensive gene expression datasets.
GSEA is a method employed to assess the enrichment trend of a specified set of genes within a gene ranking that correlates with a particular phenotype, thereby establishing the gene set's role in the manifestation of that phenotype. The analysis requires two primary inputs: a collection of genes characterized by their known functions and a matrix detailing gene expression levels. The GSEA algorithm orders genes by their degree of association with the phenotype, as reflected by variations in expression, from highest to lowest correlation. Subsequently, the software examines if the genes within each category of the gene set are clustered towards the beginning or end of the ranked list. This process reveals the influence of the collective expression changes within the gene set on the observed phenotypic differences.
GSEA was first introduced by researchers at the Broad Institute(Subramanian A.,et.al,2005).This novel strategy transitioned the analytical focus from individual genes to the collective actions of pre-defined gene sets, fundamentally establishing the basis for pathway-centric approaches within the realm of bioinformatics. Before the advent of GSEA, the examination of gene expression datasets was largely centered on detecting differentially expressed genes (DEGs) across various biological contexts, such as comparing healthy to pathological conditions. Although informative, this approach frequently failed to consider the networking of genes within biological pathways. Since diseases often entail synchronized modifications in the expression of numerous genes rather than changes in individual genes alone, investigators encountered difficulties in comprehending the complex data emanating from technologies like DNA microarrays and RNA-Seq.
Service you may intersted in
Gene sets are predefined collections of genes that are grouped together based on their association with specific biological pathways, processes, or molecular functions.
Gene sets are typically derived from various biological databases that curate and annotate genes based on their functional roles. One of the most prominent resources for gene sets is the Molecular Signatures Database (MSigDB), which contains thousands of annotated gene sets for use in GSEA(Liberzon, A.et.al). The MSigDB categorizes gene sets into several collections, including:
Unlike traditional single-gene analyses, GSEA evaluates gene sets as a whole, which reduces noise and highlights biologically relevant patterns. This provides a more holistic view of gene expression changes.
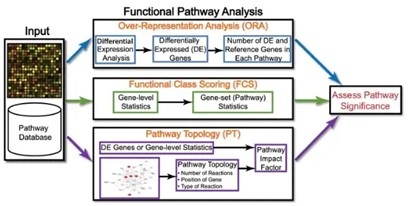 Figure1.Current gene enrichment analysis methods(Figure come from github).
Figure1.Current gene enrichment analysis methods(Figure come from github).
In the context of a gene list L that has been sequenced and a pre-established gene set S (which may include genes associated with a specific metabolic pathway, genes in close genomic proximity, or genes sharing a common Gene Ontology annotation), the objective of GSEA is to ascertain whether the genes within S are randomly scattered throughout L or are predominantly grouped at either the start or end of L. This sequencing is a reflection of the genes' varying expression levels across distinct phenotypic conditions. Should the genes within the gene set S under investigation be found to be significantly aggregated at the extremes of L, this suggests that these genes play a role in the observed phenotypic variations and are thus the focal point of our analysis.
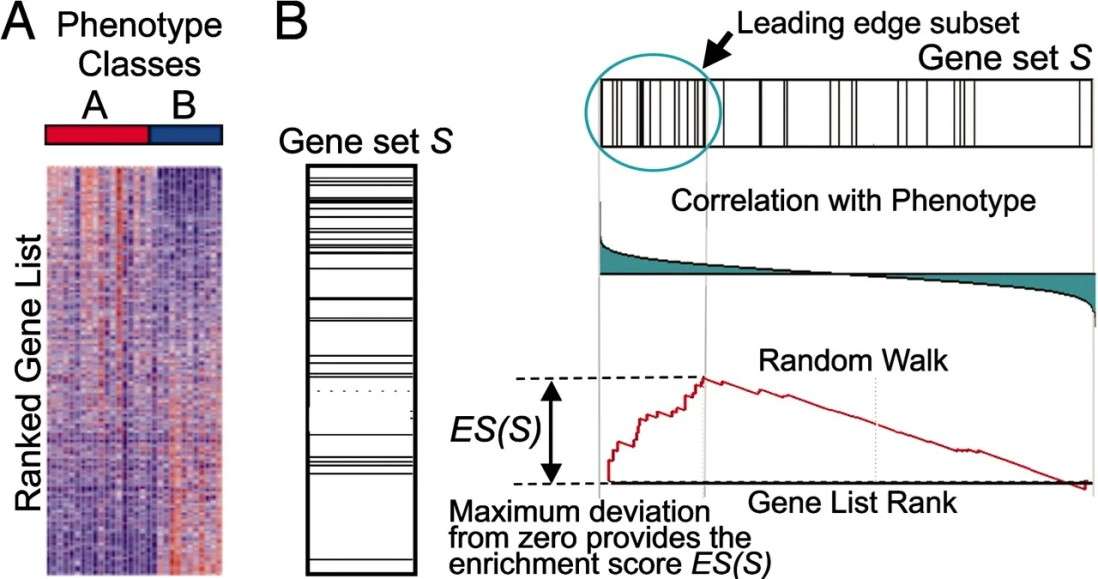 Figure2.A GSEA overview illustrating the method(Subramanian A.,et.al,2005).
Figure2.A GSEA overview illustrating the method(Subramanian A.,et.al,2005).
researchers leveraging GSEA can better interpret data from single-cell RNA sequencing and transcriptomic data analysis to reveal critical insights into cellular pathways.
Cancer Research: GSEA helps identify pathways involved in tumorigenesis, providing insights into potential therapeutic targets.
Drug Discovery: It elucidates molecular mechanisms underlying drug responses, facilitating the identification of novel drug targets.
Functional Genomics: GSEA uncovers the roles of specific gene clusters in biological processes, enhancing our understanding of gene functions.
Several tools have been developed to facilitate GSEA, each with its unique features and capabilities. Below is a table summarizing some of the most popular tools for performing GSEA, highlighting their key
| Tool Name | Description | Platform | Availability |
|---|---|---|---|
| GSEA | The original software developed by the Broad Institute for GSEA analysis. | Java-based | Open-source, free |
| Enrichr | An intuitive web-based platform that includes GSEA among other enrichment analysis methods. | Web-based | Free |
| WebGestalt | A web-based tool for gene set analysis that includes GSEA and other enrichment methods. | Web-based | Free |
| MSigDB | A collection of annotated gene sets for use with GSEA and other software. | Database | Free for academic |
| ClusterProfiler | An R package that provides various functions for gene set enrichment analysis, including GSEA. | R package | Open-source, free |
| fgsea | An R package for fast GSEA analysis that is optimized for performance with large datasets. | R package | Open-source, free |
| GSEA-MSigDB | A desktop application that integrates MSigDB with GSEA analysis. | Java-based | Free for academic |
| GenePattern | An integrated platform that includes GSEA and other bioinformatics tools. | Web-based | Free for academic |
| DAVID | A web-based tool for functional annotation and gene set enrichment analysis. | Web-based | Free for academic |
The Enrichment Score (ES) measures the degree of overrepresentation of a gene set. Other key metrics include the normalized enrichment score (NES) and adjusted p-values to ensure statistical rigor.
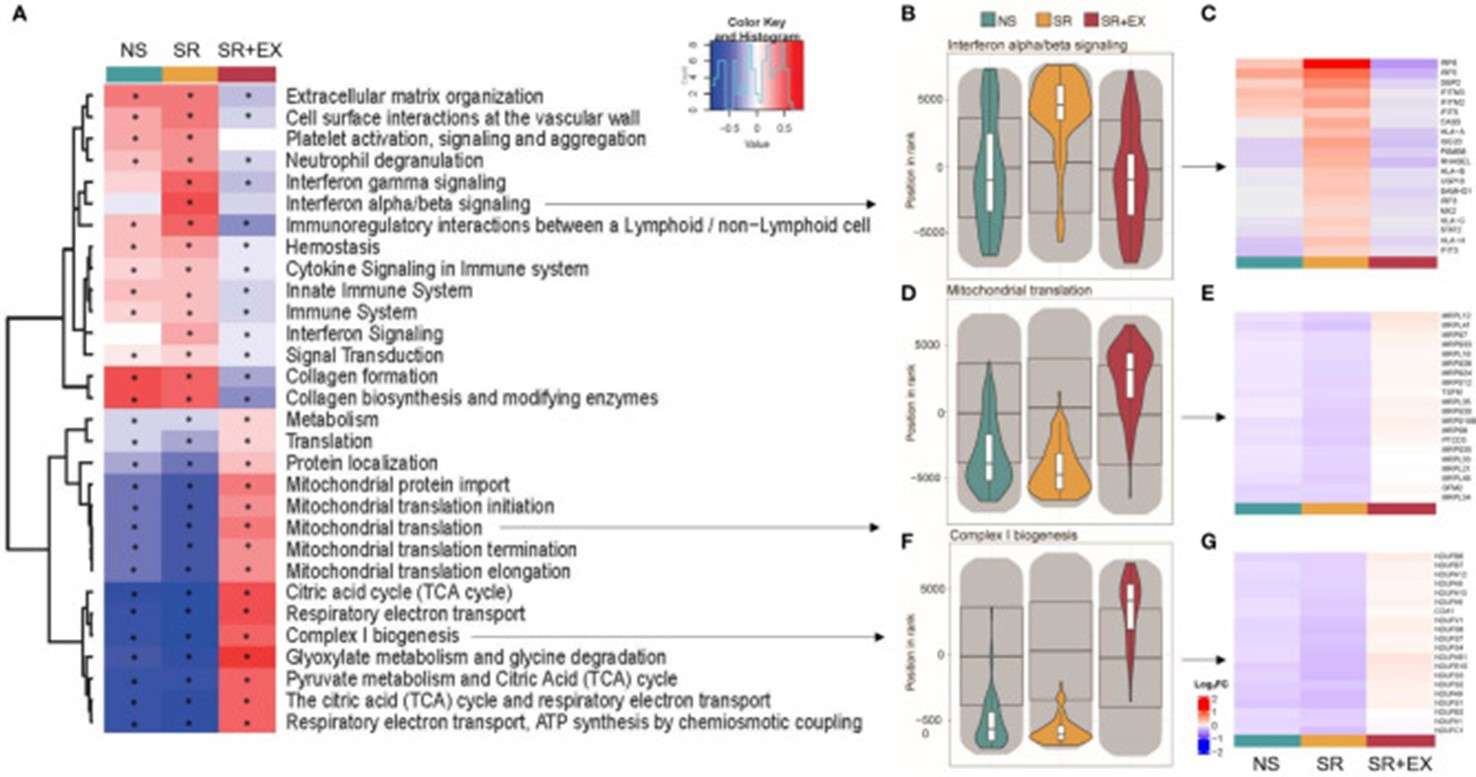 Figure3.Heatmap representing the top 30 significantly altered gene sets(Lin, W.,et.al,2022).
Figure3.Heatmap representing the top 30 significantly altered gene sets(Lin, W.,et.al,2022).
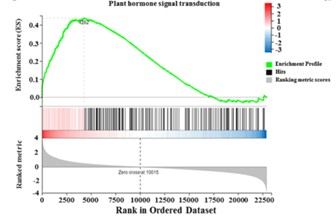 Figure4.GSEA enrichment analysis of genes related to plant hormone signal transduction pathway(Wang, Y.et.al,2021).
Figure4.GSEA enrichment analysis of genes related to plant hormone signal transduction pathway(Wang, Y.et.al,2021).
Gene Set Enrichment Analysis is a cornerstone of modern bioinformatics, empowering researchers to uncover meaningful insights from high-throughput genomic data. By leveraging robust tools and services, such as those offered by CD Genomics, researchers can enhance their understanding of gene expression and pathway dynamics.
References:


