
 Sample Submission Guidelines
Sample Submission Guidelines
Summary of Common Databases for Next Generation Sequencing
The Role of NGS Sequencing Database
In light of the accelerated advancements in technology, especially the meteoric evolution within the domain of bioinformatics, the significance of Next-Generation Sequencing (NGS) technologies as an essential instrument in life science research has markedly increased. Distinguished by its high-throughput, efficiency, and unerring accuracy, NGS offers unparalleled prospects in exploring the intricacies of life sciences. Given this context, the role of NGS sequence databases has increasingly assumed a pivotal role. These repositories have matured into an irreplaceable cornerstone within the field of life sciences, progressively shaping and expanding our comprehensive understanding of the complex tapestry that is biological systems.
NGS database stands as a comprehensive repository integrating a vast expanse of sequencing data. Within its confines, it amalgamates the diligent endeavors and research accomplishments of scientists worldwide, furnishing researchers with a rich and invaluable trove of experimental data resources. Facilitated by the NGS sequencing database, investigators gain facile access to sequencing data from diverse biological specimens, thereby enabling the unraveling of underlying biological principles and mysteries concealed within the data.
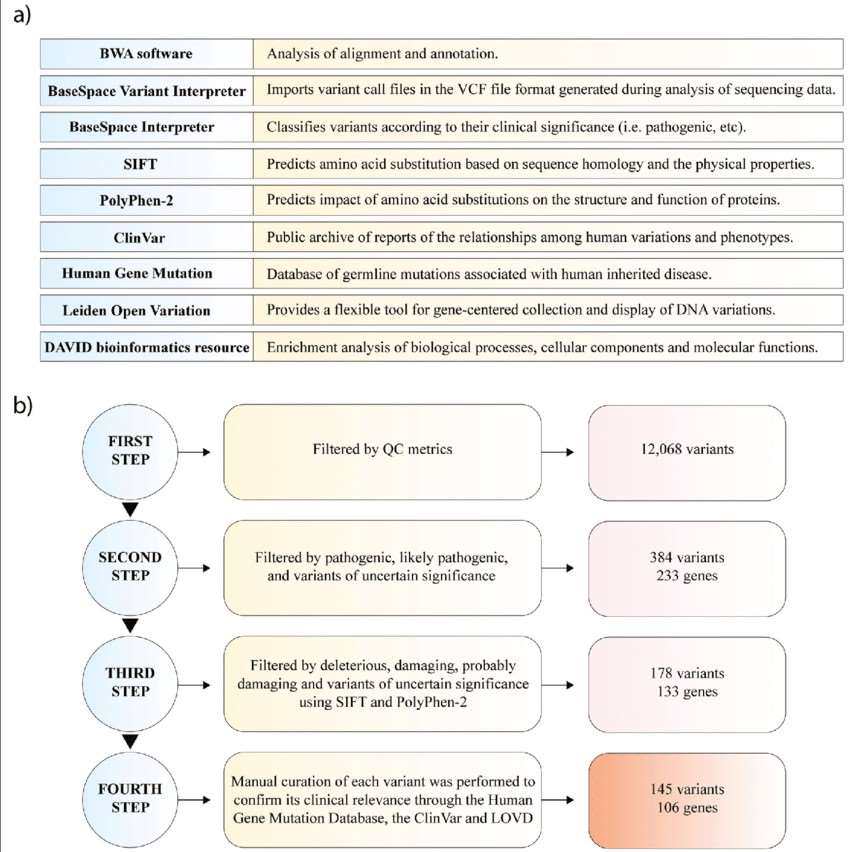 Next-generation sequencing analysis. a Functions of software and databases used for NGS analysis. b Pipeline of genomic variant analysis. (Andrés López-Cortés et al,. 2020)
Next-generation sequencing analysis. a Functions of software and databases used for NGS analysis. b Pipeline of genomic variant analysis. (Andrés López-Cortés et al,. 2020)
The role of NGS databases manifests across several dimensions:
Data Storage and Management:
NGS sequencing databases possess robust capabilities for data storage and management. They proficiently organize vast quantities of sequencing data in an efficient and structured manner, offering a multitude of convenient querying and retrieval tools that enable researchers to swiftly locate the desired data. Furthermore, these databases feature data backup and recovery mechanisms, ensuring the security and reliability of the data.
Data Analysis and Exploration:
In addition to furnishing abundant data resources, NGS databases are equipped with potent data analysis tools. Researchers harness these tools to conduct in-depth analysis and exploration of sequencing data, unveiling biological phenomena such as gene variations and expression regulation. These insights provide scientific foundations for disease diagnosis, drug development, and personalized therapies.
Data Sharing and Exchange:
Data sharing and exchange are pivotal in the realm of NGS databases, acting as expansive platforms for collaborative endeavors among researchers. Here, scientists have the opportunity to contribute their own sequencing data, thus facilitating the dissemination of their research findings to peers within the scientific community. Concurrently, researchers gain access to data contributed by others, effectively broadening their research horizons and fostering interdisciplinary collaboration. This collaborative model of data sharing and exchange is instrumental in propelling rapid advancements in life science research, driving academic collaboration and innovation forward.
Decision Support and Assistance:
NGS databases play a crucial role in decision support and assistance. They provide policymakers, clinicians, and researchers with data support regarding disease mechanisms, drug development, and treatment efficacy, aiding them in making more scientifically informed and rational decisions.
In summary, NGS sequencing databases are indispensable in life science research. They not only offer vast sequencing data resources but also encompass various functionalities such as data storage and management, data analysis and exploration, data sharing and exchange, as well as decision support and assistance. With the continuous advancement and refinement of NGS technology, the significance of NGS sequencing databases will be further accentuated, making greater contributions to the progress and innovation of life science research. This paper aims to systematically outline and elucidate the commonly used database resources in interpreting NGS sequencing reports.
You may interested in
Population Databases
RefSeqGene Database
The RefSeqGene Database, accessible at http://www.ncbi.nlm.nih.gov/refseq/rsg, stands as an openly available repository housing nucleotide sequences (DNA, RNA) along with their corresponding protein products. Conceived and developed by the National Center for Biotechnology Information (NCBI) in 2000, it retains responsibility for the curation and upkeep of this invaluable resource.
Distinguished by its taxonomic diversity, non-redundancy, meticulous annotations, and seamless integration, the RefSeq database emerges as a cornerstone in molecular research. Catering to a spectrum of scientific inquiries encompassing genomics, gene expression analyses, functional annotations, and myriad other investigative pursuits, it presents a compendium of reference sequences sourced from a broad array of organisms. This encompassing scope includes, though is not confined to, bacteria, archaea, plants, animals, fungi, and viruses.
RefSeq is a comprehensive repository housing reference sequences spanning genomes, transcripts, and proteins. Within the RefSeq database, stringent management methodologies are implemented to uphold the fidelity of gene sequences. Each sequence is accompanied by meticulous annotations, furnishing detailed insights into gene positions, exon/intron boundaries, alternative splicing isoforms, post-translational modifications, and functional domains. Drawing from a diverse array of resources, including empirical observations, computational predictions, and scholarly citations, RefSeq integrates data to offer dependable annotations for reference sequences.
Regular updates to the RefSeq database seamlessly incorporate emerging genomic, transcriptomic, and proteomic insights, guaranteeing scholars access to the most current reference sequences. This dynamic database fosters cross-referencing with other NCBI resources, such as GenBank and PubMed, empowering users to explore supplementary information and associated datasets in greater depth.
UCSC Genome Browser Database
The UCSC Genome Browser Database (https://genome.ucsc.edu/) stands as one of the most extensively utilized resources in the field of biology. Established and maintained by the University of California, Santa Cruz, it encompasses a wealth of genomic data, including gene annotation information (ENCODE), genome alignments, repetitive sequences, homologous sequences, reference sequences (mRNA, EST), phenotypes, expression profiles, regulatory information, conservation data, variations, and repetitive regions, among other information. UCSC encompasses genomic information for various common organisms, including humans, mice, fruit flies, zebrafish, nematodes, yeast, and others.
Moreover, it offers a suite of analysis tools to aid users in browsing gene information, accessing existing genome annotations, and downloading gene sequences. In the realm of bioinformatics analysis, the need for data files in formats such as fasta, GTF, or BED is ubiquitous, and UCSC serves as a primary source for accessing these files. Notably, Hg19 serves as a widely utilized reference sequence for the human genome within UCSC's repertoire.
ExAC database
(http://gnomad.broadinstitute.org)
The Exome Aggregation Consortium (ExAC) database, located at http://gnomad.broadinstitute.org, holds a central position in the genomics field. Serving as the foundational iteration of the gnomAD database, ExAC is dedicated exclusively to exome sequencing data. Its principal aim is to consolidate and standardize exome sequencing data stemming from diverse large-scale sequencing endeavors, thereby enriching the scientific community with a comprehensive array of summary data. It is noteworthy that ExAC has been seamlessly integrated into the gnomAD database, representing a notable stride forward in enhancing accessibility to genomic data and fostering collaborative research efforts.
dbSNP Database
(https://www.ncbi.nlm.nih.gov/snp/)
The dbSNP database, officially known as the Single Nucleotide Polymorphism Database, and accessible at https://www.ncbi.nlm.nih.gov/snp/, holds a pivotal position as a foundational resource curated by the National Center for Biotechnology Information (NCBI). It functions as a comprehensive repository for single nucleotide polymorphism (SNP) data spanning human genomes and certain other organisms. SNPs constitute the prevalent form of genetic variation within genomes, often originating from single nucleotide alterations that potentially contribute to inter-individual genetic diversity.
Relying on a broad spectrum of research literature, myriad databases, and multidisciplinary initiatives, the dbSNP database systematically collates and synthesizes SNP data, offering a comprehensive repository of genetic variation parameters. These include crucial specifics like SNP loci, allele prevalence, varieties of genetic variation, and intelligence germane to human health and pathology correlations. Researchers exploit the utilities of the dbSNP database to obtain detailed SNP data, corroborate SNP incidences, and explicate relationships between SNPs and particular phenotypic characteristics or pathologies.
Data housed within dbSNP are primarily categorized into two distinct types: user-submitted data, distinguished by "submitted SNP" (ss) identifiers, and data compiled from diverse submissions and additional sources, identified by "reference SNP" (rs) identifiers.
gnomAD
The Genome Aggregation Database (gnomAD), accessible at http://gnomad.broadinstitute.org, represents a collaborative effort among researchers aimed at collecting and harmonizing exome and genome sequencing data from various large-scale sequencing projects, thereby providing consolidated data to the broader scientific community. gnomAD stands as one of the most comprehensive repositories of genomic variation data to date, encompassing variants from diverse populations worldwide.
A primary distinction between gnomAD and the long-standing dbSNP database lies in their respective methodologies for cataloging genomic variations. While dbSNP aggregates variations discovered through various research methods and assigns identifiers to these variations, gnomAD employs uniform standards for the analysis of next-generation sequencing data from included samples to accurately calculate allele frequencies. This harmonization of data processing is a key characteristic of gnomAD. Additionally, gnomAD boasts high-quality data for genomic structural variations exceeding 50 base pairs.
gnomAD (v3.1.1), based on the GRCh38 reference genome assembly, encompasses a short variant dataset comprising genomic sequencing data from 76,156 unrelated individuals. This resource serves as a vital component of disease-specific investigations and population genetics research endeavors.
gnomAD (v2.1.1), based on the GRCh37 reference genome assembly, comprises a short variant dataset encompassing 125,748 exomes and 15,708 genomes. These genomic sequences originate from unrelated individuals constituting a total of 141,456 samples, forming an integral component of disease-specific investigations and population genetics research endeavors.
1000 Genomes
(https://www.internationalgenome.org/)
The aim of the 1000 Genome Project is to identify variants with a frequency greater than 1% within human populations. By sequencing a vast number of samples from diverse populations, the project has identified numerous variant sites, thereby providing a comprehensive resource for the study of human genetic variation.
Disease Databases
HGMD Database
(http://www.hgmd.org)
The Human Gene Mutation Database (HGMD) stands as the gold standard repository for genetic variants implicated in human hereditary diseases. It is meticulously curated through the comprehensive review of high-quality literature by genetic counseling experts. This database compiles and organizes information pertaining to relevant mutation sites.
HGMD provides a plethora of data associated with mutation sites, including corresponding transcripts, mutation types, pathogenicity assessments, related phenotypes, reference literature, population frequencies, functional predictions, and structural domains.
Moreover, the professional edition of HGMD offers additional details such as HGVS nomenclature for mutation sites, protein alterations, classification grades, genomic coordinates in Hg19 and Hg38 assemblies, protein structural domains, population frequencies, and software prediction outcomes.
OMIM Database
(https://www.ncbi.nlm.nih.gov/omim/)
The Online Mendelian Inheritance in Man (OMIM) database stands as a comprehensive and authoritative resource elucidating the relationship between human phenotypes and genotypes. It catalogs all known Mendelian disorders and provides information on over 16,000 genes, covering a significant portion of the human genome.
OMIM serves as a meticulous curator and integrator of published research findings, offering systematic organization and daily updates free of charge. A typical page within OMIM dedicated to a specific disease resembles a review article, comprising a spectrum of content including the relationship between phenotype and genotype, clinical synopsis, essential disease information and descriptions, clinical features, diagnosis, clinical management, pathogenesis, molecular genetics, population genetics, animal models, historical background, research progress, and references.
The ClinVar Database
(https://www.ncbi.nlm.nih.gov/clinvar/)
ClinVar serves as a publicly accessible repository housing data on human genetic variations and their correlations with diseases. It encompasses a diverse range of genetic alterations, comprising single nucleotide polymorphisms (SNPs), insertions, deletions (indels), among others. ClinVar has established itself as the foremost authoritative and comprehensive public database elucidating the associations between human genetic variations and disease phenotypes. It represents a pivotal component within the landscape of human genome information resources.
The main features of this database are as follows:
1. Data Sources: The ClinVar database primarily derives its data from a variety of literature sources, including scientific research papers, clinical diagnostic reports, and curated databases.
2. Data Format: ClinVar adopts a non-relational database structure, storing data in XML format. Data are categorized into multiple dimensions, such as variant genotypes, associations between diseases and genetic variations, variant properties, the impact of variants on host genes, and evidence.
3. Data Quality: ClinVar maintains stringent criteria for the inclusion of genetic variations, necessitating standardized representation, robust evidence support, and transparent evidence pathways. These measures ensure the accuracy and authority of the data.
InterVar Database
(http://wintervar.WGlab.org/)
The InterVar database serves as a platform for the automated assessment of pathogenicity at variant loci. It enables the automated scoring of 18 out of the 28 criteria outlined in the American College of Medical Genetics and Genomics (ACMG) guidelines. The remaining 10 criteria require additional evidence input or parameter adjustments, such as validation results from Sanger sequencing or familial validation.
ClinGen Database
(https://www.clinicalgenome.org/)
ClinGen, sponsored by the National Institutes of Health (NIH), stands as a pivotal database resource in precision medicine, facilitating the exploration of clinical relevance in genes and variations. It provides insights into the dosage sensitivity of variations, aiding in the identification of mutation visibility and recessive abnormal patterns. Moreover, ClinGen integrates clinical, genetic, population, functional evidence, and expert review to classify variations' pathogenicity based on ACMG/AMP guidelines, with results archived in ClinVar. Laboratories and clinical practitioners can utilize ClinGen's variant curation tools to assess the evidence of pathogenicity for variations that have not yet undergone expert review.
Comprehensive Cancer Database
OncoKB
(https://www.oncokb.org/)
Developed and maintained by the Memorial Sloan Kettering Cancer Center, OncoKB serves as a comprehensive oncology database centered around genetic mutations in cancer patients. It encompasses information on targeted drug usage, biological and oncological effects of mutations, as well as the distribution frequency and clinical prognostic characteristics of mutations in populations. OncoKB draws its data from diverse sources, including the FDA, NCCN, ASCO, ESMO conference papers, consensus among oncology experts, scholarly literature, as well as public databases such as cBioPortal and COSMI. Each piece of information undergoes regular review and revision by the Clinical Genomics Annotation Committee (CGAC).
Upon visiting the homepage of OncoKB, the database categorizes genetic mutation information in tumors into four data levels based on varying levels of evidence:
Level 1 data consists of mutations recommended by the FDA as biomarkers for the efficacy of drugs approved by the FDA for tumor treatment.
Level 2 data includes mutations recognized by NCCN or other expert consensus as biomarkers for the efficacy of drugs approved by the FDA for standard tumor treatment.
Level 3A data represents mutations supported by convincing clinical trial evidence as potential targets for drug treatment efficacy in tumors.
Level 3B data encompasses mutations identified through investigations indicating biomarkers for drug efficacy in standard tumor treatment, either approved or unapproved by the FDA, yet to be endorsed by expert consensus.
Level 4 data comprises mutations identified in high-quality cell or animal experimental literature as potential targets for drug treatment efficacy in tumors.
Additionally, R1/R2 level data pertains to information on mutations associated with tumor resistance: R1 denotes mutations serving as biomarkers for drug resistance indicators approved by the FDA in standard tumor treatment, while R2 pertains to mutations associated with drug-resistant tumor treatment evidence in clinical studies.
COSMIC database
(https://cancer.sanger.ac.uk/cosmic)
The COSMIC database stands as the world's largest and most comprehensive repository of somatic mutations in cancer and their implications. This resource serves as an online catalog of somatic mutations acquired in human cancers—mutations occurring in non-germline cells, thus not inherited from children. The acronym COSMIC represents the Catalogue Of Somatic Mutations In Cancer, culled from a vast array of scientific literature and large-scale experimental screenings conducted by the Sanger Institute Cancer Genome Project.
Available for free to academic researchers and licensable to others for commercial use, COSMIC currently boasts over 50,000 global users. It houses a repository of over 23,000,000 protein-coding and non-coding point mutations, 1,207,190 gene copy number variations, 19,422 fusion genes, and 7,930,489 methylation differential sites. Furthermore, the database undergoes quarterly updates to ensure relevance and accuracy.
Encompassing thousands of somatic mutations associated with cancer development, COSMIC collects data from two primary sources:
1. Known cancer gene mutations culled from literature: Genes subjected to manual curation are identified based on their presence in cancer gene census listings.
2.Data integrated from whole-genome resequencing studies of cancer samples conducted by cancer genome projects.
TCGA
(https://www.cancer.gov/ccg/research/genome-sequencing/tcga)
The Cancer Genome Atlas (TCGA), initiated jointly by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) in 2006, serves as a comprehensive repository of clinical data, genomic variations, mRNA expression, miRNA expression, methylation, and more for various human cancers, including subtypes. It stands as a pivotal resource for cancer researchers globally.
TCGA houses a multitude of data, including single nucleotide variants (SNVs), transcriptome analyses, biospecimen information, raw sequencing data, copy number variations (CNVs), DNA methylation, clinical data, among others. These data are categorized into three levels:
Level 1: Raw sequencing data (e.g., fasta, fastq files).
Level 2: Aligned bam files.
Level 3: Processed and standardized data.
| Data Type | Description |
| Clinical | Basic patient information, diagnosis, TNM staging, tumor pathology, survival status, and more |
| mRNA | mRNA expression data obtained from mRNA microarrays or RNA-seq |
| microRNA | microRNA expression data obtained from microRNA microarrays or RNA-seq |
| CopyNumber | Copy number variations of tumor segments compared to normal tissue obtained from SNP microarrays |
| Mutation | Nucleotide changes in tumor sequencing data compared to the reference genome sequence, including insertions, deletions, etc. |
| Protein | Expression levels of over 200 cancer-related proteins obtained from protein microarrays |
| Methylation | DNA methylation levels obtained from methylation microarrays |
PharmGKB
(https://www.pharmgkb.org/)
The PharmGKB database, known as the Pharmacogenetics and Pharmacogenomics Knowledge Base, epitomizes authority and comprehensiveness within the domain of pharmacogenomic databases. Established under the auspices of the National Institutes of Health (NIH), PharmGKB functions as a reservoir of insights into how human genetic variations shape responses to drugs.
Pharmacogenomics (PGx) delves into the intricate mechanisms through which genetic variations contribute to divergent responses to pharmacological agents. In an era marked by post-genomic technological advancements, pharmacogenomic research holds promise for enhancing drug efficacy while mitigating toxicity risks. Serving as a vital link between scientific exploration and clinical practice, pharmacogenomics holds immense potential for advancing the frontiers of precision medicine.
Founded in 2000, PharmGKB endeavors to curate, organize, and disseminate pharmacogenomic knowledge sourced from diverse outlets, including scholarly literature, drug labeling, and clinical directives. It occupies a central role as the primary repository for pharmacogenomic insights, encompassing drug dosing guidelines, annotations on drug labels, clinical and variant annotations, drug-centric pathways, pharmacogenomic synopses, and elucidations on the interplay between genes, drugs, and diseases.


