tRNA
tRNAs, crucial non-coding RNAs abundant in cells, hold a pivotal position in the intricate process of protein translation. These molecules act as connectors between RNA and protein codons. Notably, tRNAs undergo extensive modifications, with an average of 13 alterations per tRNA molecule. While some modifications are structural and stable, others exhibit dynamic and reversible characteristics, significantly influencing the destiny and function of individual tRNA entities.
Mutations in tRNA-modifying enzymes have been linked to a diverse spectrum of human diseases, underscoring their importance in normal cellular function. Despite their significance, there exists a notable gap in our ability to systematically and accurately quantify both the abundance of tRNAs and their modifications. A streamlined, robust, and efficient method for sequencing tRNAs is currently lacking, hindering our comprehensive understanding of these pivotal molecules and their roles in cellular processes.
Nano-tRNAseq
Recently, researchers have introduced Nano-tRNAseq, a revolutionary nanopore-based method designed for the sequencing of natural tRNAs. This innovative approach not only accurately quantifies tRNA abundance but also captures modifications, providing a robust framework for exploring the tRNAome at the single-molecule level.
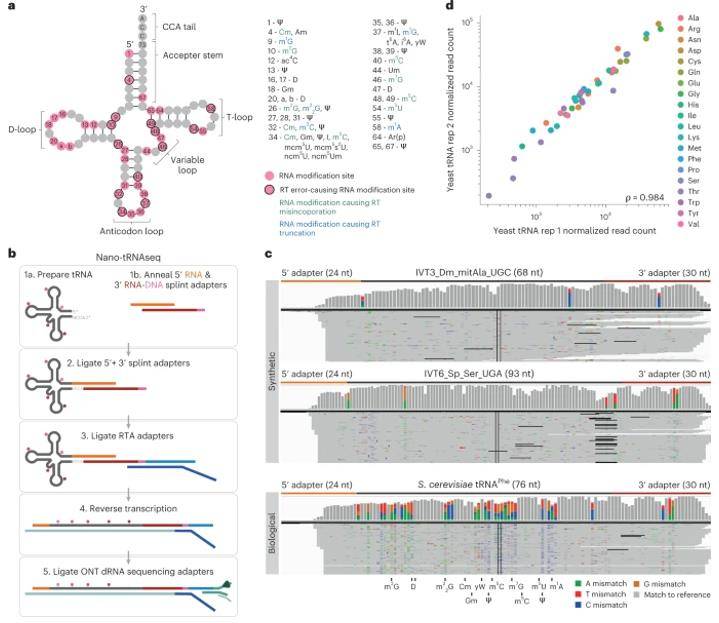 Nano-tRNAseq can efficiently sequence both IVT and native tRNA populations. (Lucas et al., 2023)
Nano-tRNAseq can efficiently sequence both IVT and native tRNA populations. (Lucas et al., 2023)
To enhance the recovery of tRNA reads, the researchers systematically reprocessed the raw nanopore current intensity signals, resulting in a remarkable 12-fold increase in the number of successfully obtained reads. This meticulous optimization enabled the faithful reproduction of accurate tRNA abundance measurements. Subsequently, Nano-tRNAseq was applied to scrutinize the Saccharomyces cerevisiae tRNA population, unveiling intricate crosstalk and interdependence among different types of tRNA modifications within the same molecule. Moreover, the method shed light on dynamic changes in the tRNA population in response to oxidative stress.
Noteworthy for its simplicity, cost-effectiveness, high throughput, and reproducibility, Nano-tRNAseq emerges as a powerful tool for investigating tRNA modifications across various biological contexts. Its versatility makes it particularly valuable for studying the biological implications of tRNA modifications in diverse scenarios such as cancer, exposure to stress, or viral infections. This breakthrough opens up exciting possibilities for utilizing tRNA molecules as potential biomarkers for monitoring human health and disease.
Nano-tRNAseq with Customized MinKNOW
Researchers have strategically employed RNA adapters to populate both the 5' and 3' ends of tRNA molecules, optimizing basecalling and mapping accuracy to capture complete tRNA sequences. Nano-tRNAseq has demonstrated exceptional success in utilizing nanopore Direct RNA Sequencing (DRS) for the comprehensive sequencing, basecalling, and mapping of both in vitro and natural tRNA molecules.
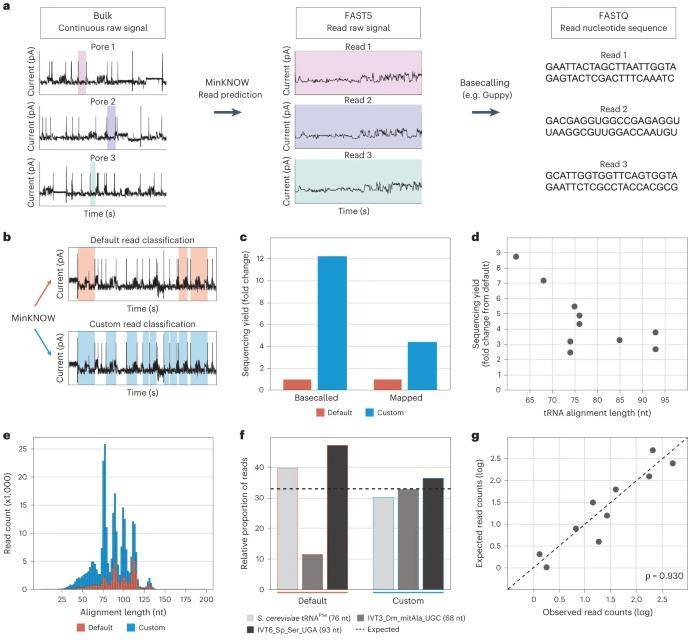 Adjustment of MinKNOW parameters increases the number of sequenced and mapped tRNA reads. (Lucas et al., 2023)
Adjustment of MinKNOW parameters increases the number of sequenced and mapped tRNA reads. (Lucas et al., 2023)
Employing linear tRNA molecules proves instrumental in mitigating pore clogging, enhancing the sequencing capacity, prolonging the integrity of the flow-through pool, and stabilizing the rate of tRNA translocation through the pore. This, in turn, refines the accuracy of the basecalling algorithm. Notably, the researchers identified that the combination of 60°C Maxima and SuperScript IV yielded optimal performance in generating full-length cDNA products, leading them to adopt 60°C Maxima for subsequent tRNA sequencing experiments.
The novel methodology introduced in this study, Nano-tRNAseq, facilitates precise and direct quantification of natural tRNA molecular abundance and their modification status using nanopore DRS. The researchers uncovered that relying solely on the double ligation of RNA adapters was inadequate for generalizing known tRNA abundances. Furthermore, the default MinKNOW configuration exhibited biased tRNA abundances, favoring longer tRNA molecules. To address these challenges, the study implemented a customized MinKNOW configuration, which not only captures tRNA reads more efficiently but also eliminates length-dependent bias, resulting in a remarkable 12-fold increase in tRNA sequencing throughput.
Reference:
- Lucas, M.C., Pryszcz, L.P., Medina, R. et al. Quantitative analysis of tRNA abundance and modifications by nanopore RNA sequencing. Nat Biotechnol (2023).
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


