




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In the field of biomedical research, the mechanism of disease occurrence and development has always been the core topic that scientists have been exploring unremittingly. In recent years, with the rise of epigenetics, DNA methylation, as a key epigenetic modification, has gradually become an important perspective for studying diseases. Reduced Representation Bisulfite Sequencing (RRBS) technology, that is, simplified representative bisulfite sequencing technology, plays an increasingly important role in analyzing the relationship between DNA methylation and diseases.
The core principle of RRBS technology is that DNA is treated by bisulfite, so that unmethylated cytosine is converted into uracil, while methylated cytosine remains unchanged. By enriching and sequencing DNA in specific regions, the methylation status of CpG sites can be accurately detected at the level of single base resolution. Compared with the whole genome bisulfite sequencing (WGBS), RRBS has obvious technical advantages, and at the same time, it can cover the key regions rich in CpG sites in the genome, such as CpG islands and promoter regions. The methylation changes in these regions are closely related to gene expression regulation and often play an important role in the process of disease occurrence.
Service you may intersted in
If you want to learn more about the RRBS, please refer to:
RRBS is widely used in tumor research, which helps to reveal the methylation regulation mechanism of cancer occurrence and development and provides targets for early diagnosis and treatment. In the field of developmental biology, we can explore the relationship between cell differentiation and dynamic changes of methylation during embryonic development. In clinical application, the detected methylation markers are helpful for disease diagnosis and prognosis evaluation, such as judging the malignant degree of tumors.
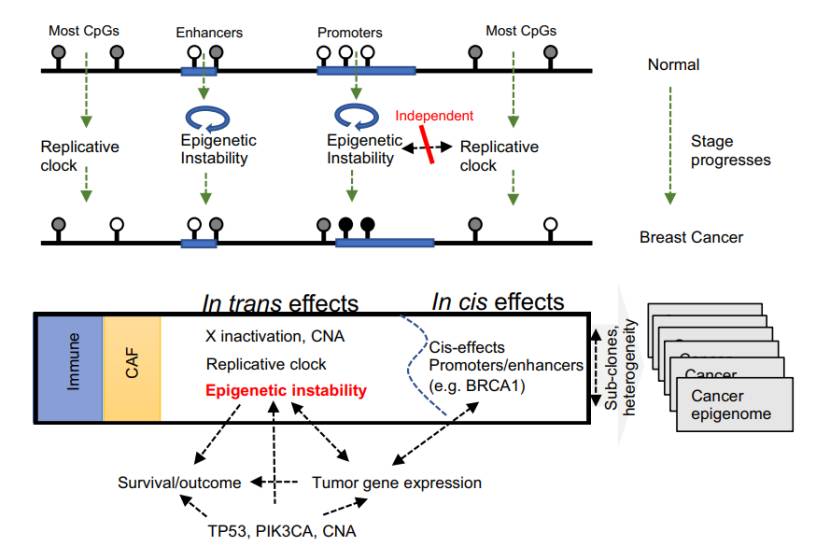 Unified model delineating the multi-factorial processes giving rise to breast cancer DNA methylation (Rajbir et al., 2021)
Unified model delineating the multi-factorial processes giving rise to breast cancer DNA methylation (Rajbir et al., 2021)
The article "Late- life exercise mitigates skeletal muscle epigenetic aging" explores the influence of exercise in old age on the epigenetic aging of skeletal muscle, and finds that exercise in old age can alleviate the epigenetic aging of skeletal muscle in mice, which provides a molecular basis for delaying muscle aging by exercise.
Background
During the whole life cycle, all tissues, including skeletal muscle, will undergo DNA methylation changes, which may lead to the decline of structure and function with age. Exercise training can change muscle DNA methylation, but it is still unclear whether it can make the skeletal muscle methylation group of old mice closer to that of young mice.
Experimental methods
The mice aged 22-24 months were trained by progressive wheel exercise (PoWeR). After training, the effects of exercise on skeletal muscle epigenetic aging were evaluated by RRBS, high-resolution targeted analysis and DNAge™ epigenetic aging clock analysis.
Main results
RRBS analysis: Compared with 4-month-old mice, skeletal muscle of 24-month-old sedentary mice has 103 unique CpG sites hypomethylation and 762 hypermethylation in the promoter region; 68 hypomethylation and 864 hypermethylation in exon region; There are 271 hypomethylation and 2261 hypermethylation in intron region. The pathway analysis of hypermethylated promoter gene showed that genes related to TCA cycle regulation were overexpressed. Exercise makes the promoter methylation state of some genes in skeletal muscle of old mice close to that of young mice, such as hypermethylation of promoter regions of Rbm10 and Timm8a1, which is relieved after exercise. RDNA is differentially methylated with age, and exercise changes the methylation status of some sites, and most unique differentially methylated rDNA sites are hypermethylated in aged exercise mice. Mitochondrial DNA(mtDNA) methylation coverage is low, and it is not affected by age or exercise.
DNAge analysis: DNAge algorithm was used to compare the epigenetic age of muscle in elderly sedentary mice and exercise mice. The results showed that the epigenetic age of PoWeR muscle was 10% lower than that in sedentary mice (about 8 weeks), and exercise reduced the variability of DNA ge estimation in elderly animals. After removing a sedentary mouse with unusually low epigenetic age, the difference of epigenetic age between exercise and sedentary aged muscles increased to about 13 weeks. The analysis of rDNAge clock shows that there is a 9% decrease after exercise, but there is no statistical significance.
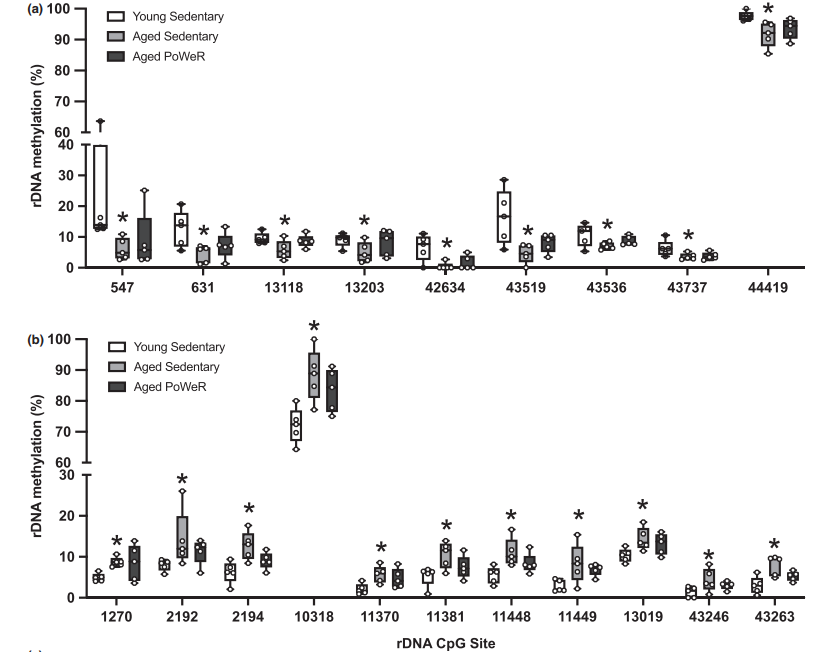 Ribosomal DNA methylation and DNAge™ analysis (Kevin et al., 2021)
Ribosomal DNA methylation and DNAge™ analysis (Kevin et al., 2021)
Conclusion
PoWeR exercise in old age can moderately but significantly reduce the hypermethylation transformation of age-related promoters, and make the epigenetic age of muscle in old mice about 8 weeks younger than that in sedentary control group, which provides a molecular basis for exercise as a therapy to alleviate skeletal muscle aging. Future research can further clarify the influence mechanism of exercise on DNAge and its relationship with age, and the epigenetic markers determined in this paper are helpful to improve muscle health in the elderly.
"DNA methylation landscapes of 1538 breast cancers reveal a replication - linked clock, Epigenomic reliability and cis-regulation" reveals the relevant characteristics and clinical significance of breast cancer methylation through DNA methylation analysis of 1538 breast cancer samples and 244 normal tissues, which provides an important basis for understanding the epigenetic mechanism of breast cancer.
Background
DNA methylation is abnormal in cancer, but its related dynamics, regulation and clinical significance are unknown. The genome of breast cancer is affected by copy number variation and a few gene mutations, and the early research on DNA methylation is limited, so it is necessary to comprehensively analyze the relationship between the regulatory layers.
Experimental method
1538 tumors and 244 normal tissue samples in METABRIC cohort were sequenced by RRBS, and DNA methylation was analyzed. The average coverage was 3.3M CpGs, and most samples had good CpG coverage.
Methylayer calculation strategy was developed to integrate gene expression, genetic and clinical information, analyze tumor methylation dynamics, screen cis-regulatory candidate genes and prognostic indicators, and verify them in independent data sets.
Main results
Analysis of methylation characteristics: It was found that the methylation characteristics related to immunity and cancer-associated fibroblasts (CAF) in tumor microenvironment (TME) were related to tumor grade; The methylation lost clock related to tumor replication was identified, mainly in the late replication region, which was less related to gene expression. Two characteristics of epigenetic instability, namely CpG island methylation gain (MG) and loss (ML), were identified, which were related to tumor progression, gene expression and genome subtype.
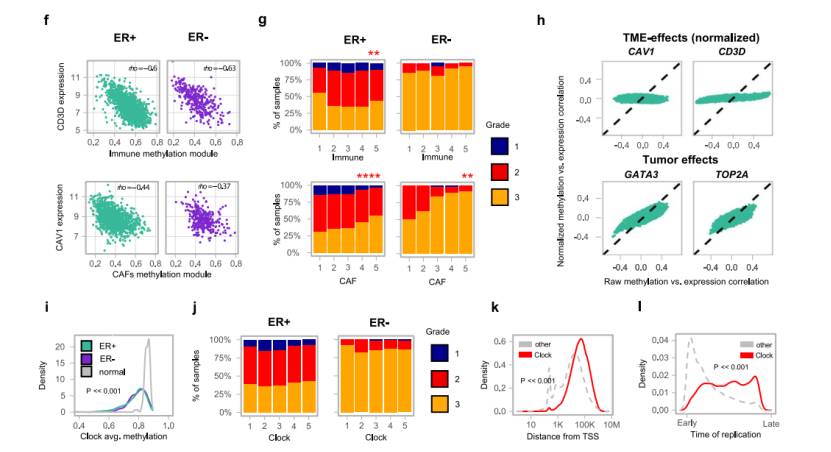 A replication-linked methylation clock process correlates with pervasive loss of methylation in tumors(Rajbir et al., 2021)
A replication-linked methylation clock process correlates with pervasive loss of methylation in tumors(Rajbir et al., 2021)
Cis-regulatory relationship: Cis-regulatory relationship between methylation and expression was found in hundreds of promoters and remote elements. These regulatory relationships are gene-specific, and the methylation diversity of some gene promoters and remote elements decreased, suggesting a selective role in the process of tumorigenesis.
X-linked dose compensation: X-chromosome dose compensation is related to methylation, especially in ER-tumor, and there is also local dose compensation at non-X-linked sites.
Clinical relevance: Methylation characteristics are related to genomic variation, and MG and ML are related to poor survival rate, which can provide information for the prognosis of breast cancer.
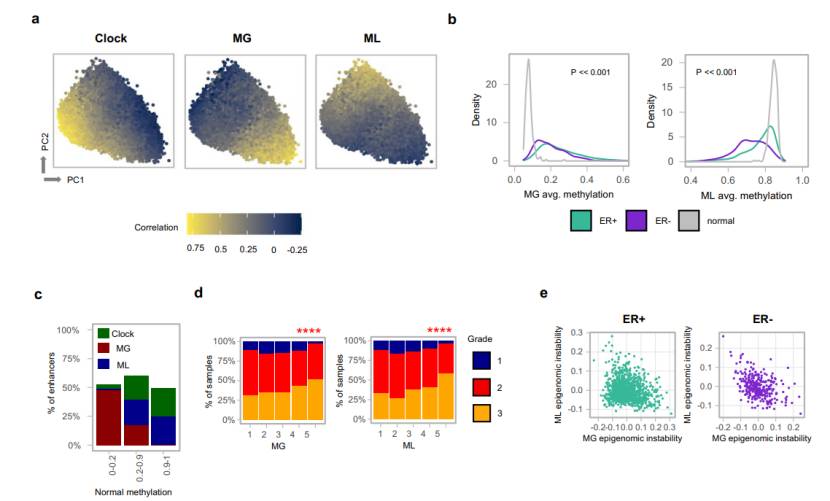 Epigenomic instability in breast cancers (Rajbir et al., 2021)
Epigenomic instability in breast cancers (Rajbir et al., 2021)
Conclusion
A multi-factor model of DNA methylation in breast cancer was established, which showed that epigenetic instability was an important feature of breast cancer, and it was related to tumor gene expression, progress and prognosis, which provided a new perspective for disease evaluation and treatment strategy.
The article "DNA methylation signatures of Alzheimer's disease neuropathology in the cortex is primaryly driven by variation in non-neuronal cell-types" is based on the analysis of large and medium-sized cells. By analyzing the DNA methylation of samples and exploring the epigenetic characteristics of Alzheimer's disease (AD), it is found that most differential methylation sites in cortex reflect the DNA methylation differences of non-neuronal cells, which provides important clues for AD research.
Background
AD is a chronic neurodegenerative disease, and its molecular mechanism is unknown. The existing researches on the epigenetics of AD have some limitations, such as it is difficult to solve the problem of cellular heterogeneity in cortical tissue, and it has not comprehensively analyzed various pathological indexes. The purpose of this study is to reveal the characteristics of DNA methylation related to AD by using multi-pathological measurement and cell purification techniques.
Experimental method
The cerebral cortex (DLPFC and OCC regions) of 631 donors were sequenced for DNA methylation, and the differential methylation sites (DMPs) related to AD pathology were analyzed, and the factors such as age and sex were controlled.
A cell type deconvolution model based on NeuN+, SOX10+ and neun–/sox10–nuclei was developed to estimate the cell proportion and analyze its relationship with AD pathology.
The DNA methylation of some donors' purified nuclear populations was analyzed and compared with the results of bulk tissue.
The data of this study and previous studies on DNA methylation in AD cortex were integrated for meta-analysis.
Main results
Cell proportion is related to AD pathology: in DLPFC, the pathological increase of tau and amyloid protein is related to the decrease of NeuN+ cell proportion, the increase of SOX10+ cell proportion and the decrease of neun–/sox10–cell proportion, but not in OCC.
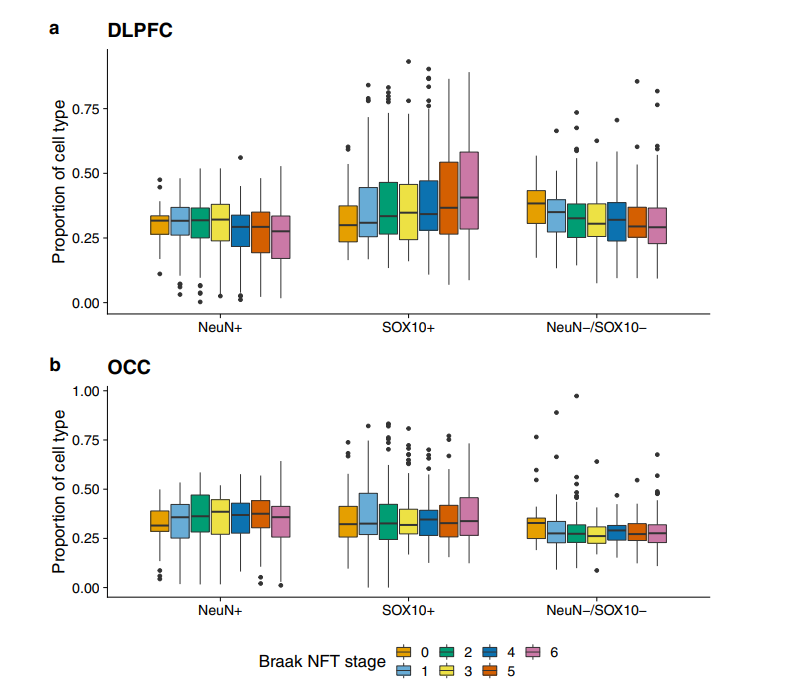 DNA methylation data in the DLPFC (Gemma et al., 2022)
DNA methylation data in the DLPFC (Gemma et al., 2022)
Multiple DMPs were identified: DMPs related to AD pathology (tau, amyloid, LB and TDP-43 pathology) were identified in cortex, such as cg06913337(ZFPM1 gene). Meta-analysis combined with the data of 2013 donors identified 334 DMPs related to tau pathology, of which 140 were new sites, involving immune, inflammatory, mitochondrial function and other pathways.
DMPs is different Brain region And the characteristics of cell types: the difference of DNA methylation related to AD is highly consistent in DLPFC and OCC. The analysis of purified nuclei showed that most DMPs in bulk cortical tissue reflected the difference of DNA methylation in non-neuronal cells (especially NeuN-/SOX10- cells), and the effect amount increased greatly in these cells.
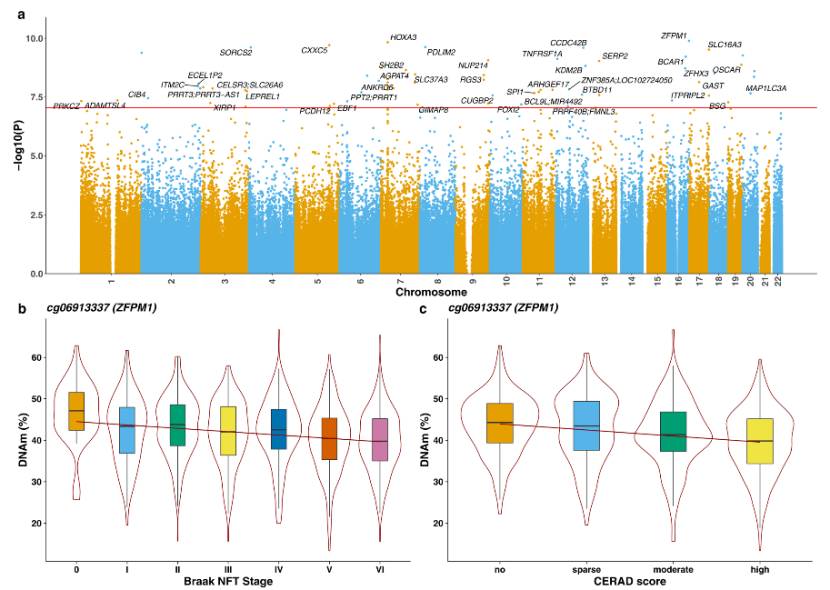 DMPs in the cortex associated with Alzheimer's disease neuropathology (Gemma et al., 2022)
DMPs in the cortex associated with Alzheimer's disease neuropathology (Gemma et al., 2022)
Conclusion
In this study, multiple DMPs related to AD pathology were identified by using multi-pathological measurement and cell purification techniques, and it was found that most DMPs in cortex originated from methylation differences of non-neuronal cells. The importance of multi-pathological measurement and cell type analysis in the epigenetic study of AD was emphasized, which provided a basis for further understanding the pathogenesis of AD.
Paper "Integrated DNA methylation and transcript omics analysis of lactic Glands identify the potential genes implied in the development of Sjgren's syndrome-related dry eye" was written by Mei Sun et al. The etiology of SS-related dry eye is unknown, and the role of epigenetic factors is concerned, but the DNA methylation of lacrimal gland is unclear. In this study, NOD mice were used to analyze the changes of DNA methylation and transcriptome in lacrimal gland by using RRBS and RNA-Seq techniques, and to explore the relationship between DNA methylation and diseases, so as to provide targets for diagnosis and treatment.
Background
The non-obese diabetic (NOD) mice, which spontaneously develop ocular surface dryness and autoimmune dacryoadenitis that resemble human SS-related dry eye, have been widely used in SS dry eye-related studies. For example, some researches demonstrated that treatment with mesenchymal stem cells could ameliorate the clinical manifestations of SSrelated dry eye in NOD mice by increasing tear secretion, decreasing corneal stain scores, recovering goblet cell counts in the conjunctiva, and attenuating foci inffltrations in the LGs.
Experimental method
Characteristics of disease progression in animal model: NOD mice aged 4-16 weeks were used for research, and the disease was evaluated by corneal fluorescein staining, tear secretion test and lacrimal gland histology.
Changes of DNA methylation in lacrimal gland: The RRBS sequencing of lacrimal gland of NOD mice aged 4-16 weeks showed that the DNA methylation level of lacrimal gland of 8, 12 and 16 weeks old mice was higher than that of 4 weeks old, mainly at CG site and the methylation of promoter region changed, suggesting that it was related to the pathogenesis of the disease, and Dnmt1 and Dnmt3b might play a major regulatory role.
Transcriptome analysis of lacrimal gland: RNA-Seq analysis showed that there were 1321, 2549 and 3712 differentially expressed genes (DEGs) in lacrimal gland of NOD mice at 8, 12 and 16 weeks of age, which were mainly involved in immune response-related processes, such as T cell activation and MHC II protein complex binding. KEGG analysis showed that the related pathways were enriched, indicating that immune response-related genes were up-regulated.
Correlation analysis between methylation and gene expression: Focusing on gene promoter region DMRs, NOD mice at 8, 12 and 16 weeks old have 140 promoters with hypomethylation and up-regulated genes, and their functions are enriched in T cell proliferation and activation.
Main results
NOD mice showed corneal epithelial defects, decreased tear secretion and lymphocyte infiltration from 8 weeks old, and the symptoms were obvious from 12 to 16 weeks with age, indicating that SS-related dry eyes appeared at 8 weeks and gradually deteriorated.
The differential methylation regions (DMRs) of mice at different ages were calculated. There were 4132, 2396 and 26135 DMRs at 8, 12 and 16 weeks, respectively. The functions of related genes (DMGs) were enriched in GTPase activation, Ras signaling pathway and so on, which involved immune cell migration and dry eye disease development.
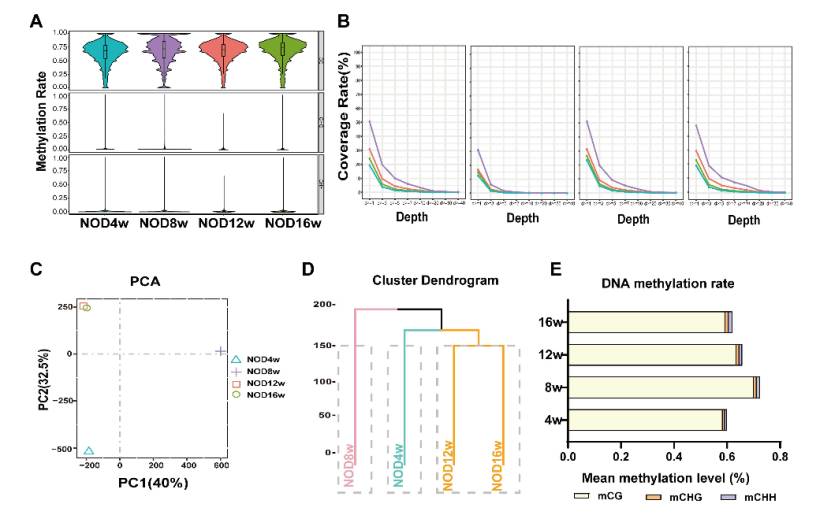 DNA methylation levels were increased in LGs of NOD mice (Sun et al., 2023)
DNA methylation levels were increased in LGs of NOD mice (Sun et al., 2023)
Eight key genes (Icosl, Batf, Irf4, Itgal, Vav1, Tgfβ1, Jak2 and CD86) were screened out. Among them, the expressions of Icosl, Irf4, Vav1 and Itgal are positively correlated with inflammatory cell infiltration, and the promoter is hypomethylated, which indicates that they participate in disease development by regulating T cell response.
Conclusion
This study shows that the DNA methylation changes of lacrimal gland in the development of SS-related dry eye in NOD mice are related to T cell-related genes, which provide a target for etiology and treatment. However, the samples are from mice and need to be verified in patients, and the related genes, pathway mechanisms and cell heterogeneity need further study
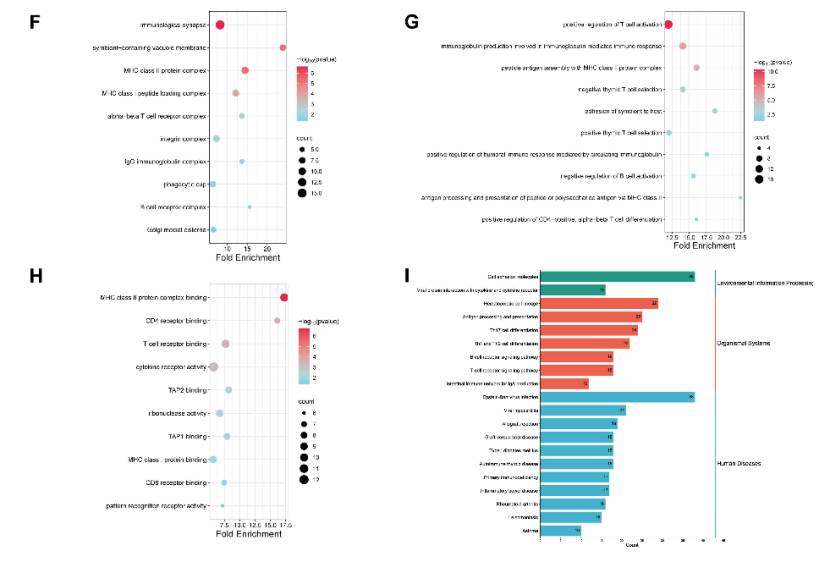 Transcriptome analysis of LGs during the development of SS-related dry eye (Sun et al., 2023)
Transcriptome analysis of LGs during the development of SS-related dry eye (Sun et al., 2023)
Although RRBS technology has made remarkable progress in disease research, it still faces some challenges. For example, data processing and analysis are complex, which requires powerful bioinformatics tools and professional knowledge to interpret massive sequencing data and accurately identify differential methylation sites and regions that are truly related to diseases. The selection and processing of samples also need strict standardized processes to ensure the reliability and repeatability of the results.
Looking forward to the future, with the continuous innovation and improvement of technology, RRBS technology is expected to play a more critical role in disease research. Combined with multi-omics technologies, such as transcriptomics and protein omics, we can more comprehensively analyze the regulatory network of DNA methylation in the occurrence and development of diseases, provide a more solid foundation for the development of precision medicine, help mankind overcome more disease problems and improve health level.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.