DAP-seq vs ChIP-seq:Comparison of Protein-DNA Interactions in the Genome
Transcription factors (TFs) are instrumental regulatory proteins that intricately modulate gene expression levels by engaging with specific binding sites termed TF binding sites (TFBS) nestled within gene promoter regions. Unveiling the multifaceted roles of TFs and their interactions with target genes serves as a cornerstone in unraveling the intricate machinery of gene regulation. Recent strides in high-throughput technologies have propelled significant advancements in this domain. Breakthroughs such as chromatin immunoprecipitation sequencing (ChIP-seq) and DNA affinity purification sequencing (DAP-seq) have emerged as robust methodologies for precisely mapping TF binding sites across the genome. The fusion of these state-of-the-art approaches has substantially augmented our understanding of both the structural framework and operational dynamics of gene regulatory networks. Although DAP-seq and ChIP-seq technologies both serve as indispensable tools for probing protein-DNA interactions and pinpointing transcription factor binding sites, they diverge in their foundational principles, procedural workflows, and real-world applications. This article aims to undertake a comprehensive comparative analysis delineating the disparities between these two techniques across various dimensions.
what is DAP-seq?
The Principles and workflow of DAP-seq Technology
DAP-seq is a highly efficient method developed by the team led by Joseph R. Ecker at the Salk Institute in the United States in 2016, aimed at identifying TF binding sites ex vivo. This technique ingeniously combines protein expression technology outside of cells with high-throughput sequencing, enabling us to utilize externally expressed TFs to bind to genomic DNA without the need for specific antibodies for each TF. This innovation renders DAP-seq an ideal choice for studying TF binding site information in non-model plants. In the process of DAP-seq, TFs are first allowed to form complexes with DNA through ex vivo experiments, followed by enrichment of DNA fragments bound by TFs using DNA affinity purification technology. Subsequently, these DNA fragments are eluted and subjected to high-throughput sequencing and analysis, thereby determining the binding sites of TFs in the genome and generating a comprehensive map of TF binding sites across the entire genome. The significant advantage of DAP-seq lies in its high throughput and low background noise, making it an effective tool for studying TF binding sites.
In Vitro Protein Expression
Affinity Purification and Sequencing
- Library Construction: The process initiates with the extraction of genomic DNA from samples, followed by fragmentation into approximately 200 bp fragments. After repair, these fragments are ligated with sequencing adapters using Illumina technology, facilitating the construction of a genomic DNA library.
- In Vitro Protein Expression: Subsequently, coding sequences (CDS) encoding the desired TFs are inserted into vectors containing an affinity tag, such as the Halo Tag, to generate protein expression vectors. Protein expression is then conducted under controlled in vitro conditions, where synthesized proteins conjugate with ligand-bound magnetic beads. This strategic coupling eliminates non-specific proteins, resulting in fusion proteins comprising TFs and the affinity tag.
- Affinity Purification and Sequencing: After protein expression, purified fusion proteins and the affinity tag incubate with the genomic DNA library. Leveraging specific binding affinity, complexes form with certain genomic DNA sequences. These complexes undergo extraction and purification using Halo Tag-specific magnetic beads, removing unbound DNA fragments. Captured DNA fragments are finally PCR-amplified and sequenced using the advanced Illumina platform.
- Data Analysis: Sequenced reads are meticulously aligned with the reference genome, facilitating identification of potential TF binding sites.
The advantages and limitations of DAP-seq
DAP-seq stands out for its ability to provide high-resolution information on transcription factor binding sites. What sets it apart is its independence from specific antibodies, mitigating experimental reliance and the risk of nonspecific binding issues. Covering the entire genome, DAP-seq accurately identifies TF binding sites, offering comprehensive insights for research. Moreover, its versatility extends beyond TFs, allowing for the study of various DNA-binding proteins, broadening its molecular research applications. However, despite its high resolution, DAP-seq may encounter false positives and negatives, especially in detecting low-abundance binding sites, leading to variable success rates across different TF families. Additionally, the substantial data output requires sophisticated bioinformatics tools and expertise for analysis and interpretation. Optimizations may also be necessary for different proteins and biological samples to achieve optimal binding site detection.
The Application of DAP-seq in Genomics and Biological Research
The application of DAP-seq technology in genomics and biological research is extensive. Firstly, DAP-seq accurately identifies TFBS, thereby revealing the mechanisms and biological processes of gene expression regulation. For instance, researchers have utilized DAP-seq to identify the binding motif of an AP2/ERF TF and its target genes in maize. This revealed how ZmEREB57 regulates maize OPDA synthesis through two distinct signaling pathways, enhancing salt tolerance and providing crucial genetic resources for breeding salt-tolerant plant varieties. Additionally, DAP-seq analysis can delve into the impact of DNA methylation on TF binding sensitivity, elucidating the role of epigenetics in gene expression regulation. For example, researchers have constructed a genome-wide TFBS map in Arabidopsis using DAP-seq, uncovering the influence of methylation on TFBS. Moreover, by integrating DAP-seq technology with multi-omics analysis, researchers can reshape TF regulatory networks, gaining deeper insights into important biological processes such as growth, development, and environmental adaptation.
what is CHIP-seq?
The Principles and workflow of DAP-seq Technology
ChIP-seq combines ChIP technology with next-generation sequencing to efficiently detect DNA segments involved in interactions with histones, TFs, and other proteins across the genome. The principle involves first selectively enriching DNA fragments bound by the target protein using ChIP and subsequently purifying and constructing libraries from these fragments. Then, high-throughput sequencing is performed on the enriched DNA fragments. By precisely mapping millions of obtained sequence tags to the genome, researchers gain insights into DNA segments interacting with histones, TFs, and other proteins across the entire genome.
- Sample Preparation: Prepare cells containing the protein that you are interested in. These cells are meticulously sourced from diverse origins, ranging from cell cultures to animal tissues or clinical samples. Throughout this preparatory phase, utmost attention is devoted to preserving the biological characteristics and structural integrity of these cells. Such diligence is imperative to ensure the reliability and accuracy of subsequent analyses and interpretations.
- Cross-linking Fixation: Upon acquiring the cells, we embark on the process of cross-linking fixation to immobilize proteins and DNA within the intricate cellular environment. Employing cross-linking agents such as formaldehyde, this step enables the capture of direct protein-DNA interactions while maintaining their native conformation. Preserving the structural integrity of these interactions is crucial for faithfully representing the biological context within the cellular milieu.
- Cell Lysis: Following fixation, cells gracefully yield to the process of lysis, liberating chromatin and proteins from the cellular matrix. Leveraging a harmonious blend of chemical and physical methodologies such as sonication or enzymatic treatment, cell structures are dismantled, facilitating the release of cellular constituents essential for downstream analyses.
- Immunoprecipitation: Specific antibodies are then deployed as molecular sentinels to selectively bind the target protein onto solid-phase substrates such as magnetic or agarose beads. These antibodies demonstrate remarkable specificity, recognizing and capturing target proteins with precision, thus forming protein-DNA complexes pivotal for subsequent investigations.
- Cross-link Reversal: The subsequent critical phase involves delicately reversing the cross-linking agents through controlled heating or chemical treatment. This meticulous process disentangles proteins from DNA, reinstating DNA to its original state and ensuring the fidelity of subsequent analyses.
- DNA Extraction: From the intricate network of immunoprecipitated complexes, DNA extraction emerges as a pivotal endeavor. Leveraging DNA extraction kits or alternative methodologies, purified DNA is meticulously extracted, ensuring its freedom from potential contaminants, thereby laying the foundation for downstream analyses.
- Library Construction and Sequencing: Extracte DNA fragments undergo a transformative journey encompassing end repair, adapter ligation, and polyA tail addition to construct DNA libraries. Subsequently, we harness high-throughput sequencing technologies such as Illumina sequencing to unveil a vast array of short-read sequences, thereby illuminating the genomic landscape in intricate detail.
- Data Analysis: The culmination of our experimental odyssey involves exhaustive processing and analysis of the obtained sequencing data. From meticulous quality control to intricate motif analysis, each analytical endeavor aims to unravel the complexities of protein-DNA interactions, shedding light on specific binding sites and elucidating the biological significance encoded within the genome.
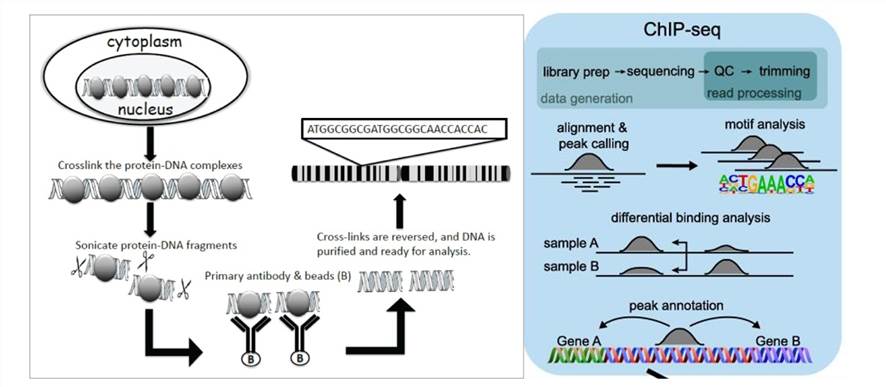 Principle of ChIP assay and ChIP-seq(Mundade et al., 2014; Höllbacher et al.,2020)
Principle of ChIP assay and ChIP-seq(Mundade et al., 2014; Höllbacher et al.,2020)
Service you may intersted in
The advantages and limitations of ChIP-seq
ChIP-seq stands as a cornerstone in genomics, offering a panoramic insight into protein-DNA interactions across the genome. This technique empowers researchers to pinpoint binding sites for various DNA-binding proteins like TFs and histones, unraveling the complexity of gene regulation mechanisms. Moreover, its dynamic nature allows exploration of how these interactions evolve under different biological conditions, providing invaluable insights into gene regulation nuances.
However, ChIP-seq faces notable limitations. Primarily, it relies on specific antibodies tailored to the protein of interest, which can be challenging to obtain or may lack the desired specificity, impacting analysis accuracy. Furthermore, its intricate data analysis requires a deep understanding of bioinformatics and substantial computational resources. Additionally, susceptibility to background noise and false positives, especially in regions with low DNA-binding affinities, presents a hurdle. Lastly, the technical complexities demand meticulous optimization for varied cell types and conditions, potentially introducing bias into findings.
The Application of ChIP-seq in Genomics and Biological Research
ChIP-seq emerges as a pivotal tool in deciphering the intricate landscape of protein-DNA interactions and epigenetic regulation within the genome. Through ChIP-seq, the precise delineation of TFBS across the genome facilitates the revelation of gene regulatory mechanisms and expression patterns. Furthermore, ChIP-seq enables exploration into DNA methylation patterns, as well as the investigation of histone modifications and nucleosome positioning, thus advancing our understanding of chromatin architecture and function. For instance, the development of ChIP-seq-based centromere isolation assays utilizing CENH3 antibodies has enabled the separation of centromere DNA from different species, propelling in-depth investigations into centromere biology. Additionally, leveraging ChIP-seq technology, researchers have successfully identified a series of centromere-specific repeat sequences in citrus, unveiling valuable insights into centromere evolution through comprehensive bioinformatic analyses and the discovery of G-quadruplex structures within citrus centromere repeat sequences, thereby catalyzing further exploration into centromere origins, evolution, and genomic studies.
Comparison between DAP-seq and ChIP-seq
| Feature |
DAP-seq |
ChIP-seq |
| Principle |
Utilizes the protein's inherent DNA affinity for purification and sequencing |
Relies on specific antibodies to immunoprecipitate protein-DNA complexes |
| Antibody Dependency |
Independent; does not require antibodies, reducing the risk of non-specific binding |
Dependent; requires high-quality, specific antibodies for successful immunoprecipitation |
| Resolution |
Typically offers higher resolution, allowing for more precise localization of binding sites |
Lower resolution compared to DAP-seq; may require optimization for increased precision |
| Sample Preparation |
Requires high-quality and well-purified samples |
Requires effective cross-linking, antibody selection, and immunoprecipitation steps |
| Data Analysis |
Requires complex bioinformatics tools for data processing and interpretation |
Also requires advanced data analysis, with additional considerations for antibody selection and validation |
| Application Scope |
Suitable for a variety of DNA-binding proteins |
Primarily used for studying the DNA binding characteristics of specific proteins |
| False Positives/Negatives |
High resolution makes it more sensitive to low-abundance binding sites |
Potential for false positives due to non-specific antibody binding |
| Cost |
Can be costly, especially in sample preparation and data analysis |
Also expensive, with significant costs associated with antibody quality and validation |
| Advantages |
High resolution, no reliance on antibodies, compatibility with diverse omics data |
Well-established technique, widely applied in various biological research areas |
DAP-seq and ChIP-seq emerge as dual engines of discovery in the realm of genomics, adept at pinpointing the precise locations where TFs anchor to the DNA, thereby forecasting their downstream regulatory effects.DAP-seq excels in vitro, dissecting the protein-DNA dialogue in a controlled environment that transcends species boundaries, making it a versatile choice for uncovering the genomic binding blueprint of proteins, including but not limited to TFs. This technique's independence from antibodies simplifies the process, especially for plant research where antibody acquisition can be a formidable hurdle.Conversely, ChIP-seq operates in vivo, offering an authentic glimpse into the dynamic interplay between proteins and DNA within the cellular milieu. When the research zeroes in on a specific protein for which high-fidelity antibodies are at hand, ChIP-seq becomes the method of choice, capturing the native state of protein-DNA associations.
Ultimately, the decision to employ DAP-seq or ChIP-seq rests on the research's specific objectives, the resources at disposal, and the characteristics of the protein under the microscope. As the landscape of genomics research continues to expand, we stand on the cusp of methodological breakthroughs that will sharpen our understanding of the complex choreography between proteins and DNA, further enriching our biological insights.
References
-
Bartlett A, O'Malley RC, Huang SC, et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc. 2017;12(8):1659-1672. doi:10.1038/nprot.2017.055
- Mundade R, Ozer HG, Wei H, Prabhu L, Lu T. Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond. Cell Cycle. 2014;13(18):2847-2852. doi:10.4161/15384101.2014.949201
- Höllbacher B, Balázs K, Heinig M, Uhlenhaut NH. Seq-ing answers: Current data integration approaches to uncover mechanisms of transcriptional regulation. Comput Struct Biotechnol J. 2020;18:1330-1341. Published 2020 May 31. doi:10.1016/j.csbj.2020.05.018
- Zhu J, Wei X, Yin C, et al. ZmEREB57 regulates OPDA synthesis and enhances salt stress tolerance through two distinct signalling pathways in Zea mays. Plant Cell Environ. 2023;46(9):2867-2883. doi:10.1111/pce.14644
! For research purposes only, not intended for clinical
diagnosis, treatment, or individual health assessments.



