




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Epigenetic modifications function as regulatory mechanisms that exert precise control over chromosomal DNA and associated proteins through chemical alterations. These modifications influence various genetic processes, including transcription, splicing, translation, nucleosome assembly, and chromatin structure. Such mechanisms are vital in plant growth and development and significantly impact responses to biotic and abiotic stressors. Among the diverse forms of epigenetic modifications, DNA modifications, histone modifications, and chromatin remodeling are particularly critical.
This study centers on DNA modifications, with a specific focus on DNA methylation, a key modification mechanism. DNA methylation, an integral component of DNA modifications, is intricately linked to essential biological processes such as gene expression, growth, development, and stress responses in plants. The investigation of DNA methylation has become a prominent area of research within the field of epigenetics.
Whole-genome bisulfite sequencing (WGBS/BS-seq) constitutes the gold standard for the investigation of DNA methylation attributable to its superior advantages. This technique enables a thorough and precise examination of plant DNA methylation, employing highly efficient and accurate methodologies. The primary aim of this paper is to elucidate the fundamental principles underlying DNA methylation and to delineate the specific applications of BS-seq technology within the context of plant DNA methylation research. This discourse intends to furnish valuable insights and references for researchers specializing in related domains.
DNA methylation, an integral form of epigenetic modification, is catalyzed by DNA methyltransferases. This biochemical process entails the covalent attachment of a methyl group to the DNA molecule, utilizing S-adenosylmethionine (SAM) as the methyl donor (Lister et al., 2008). DNA methylation exhibits site-specificity, predominantly targeting the C-5 position of cytosine, N-6 position of adenine, and G-7 position of guanine residues. In empirical research, the primary focus often lies on the methylation of the fifth carbon of cytosine within CpG dinucleotide sequences, resulting in the formation of 5-methylcytosine (5-mC). This methylation pattern represents the predominant form of DNA methylation in eukaryotic organisms, encompassing both plants and animals.
For more information about DNA Methylation, please refer to What is DNA Methylation.
In plants, DNA methylation entails dynamic regulation through three primary processes: de novo methylation, maintenance methylation, and demethylation, each catalyzed by distinct enzymes (Figure 1).
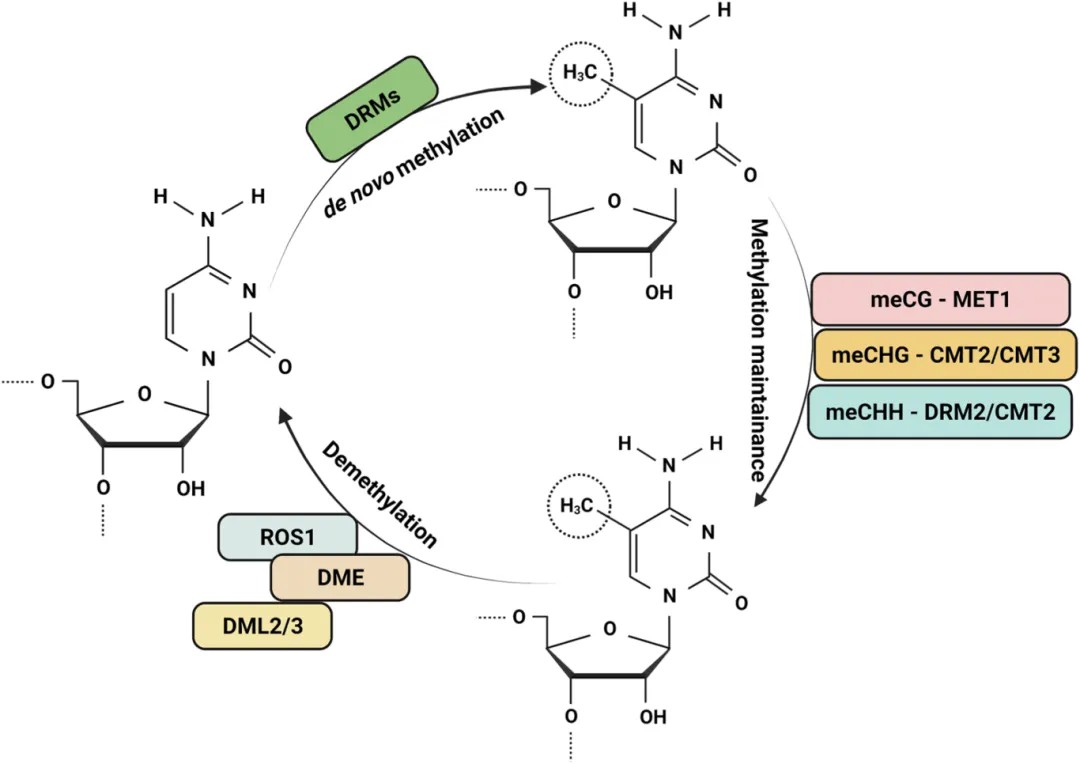 Figure 1. Dynamic regulation of DNA methylation and related enzymes in plants (Arora et al., 2022).
Figure 1. Dynamic regulation of DNA methylation and related enzymes in plants (Arora et al., 2022).
De novo methylation in plants occurs through the RNA-directed DNA methylation (RdDM) pathway, as elucidated by Matzke et al. in 2014. The RdDM pathway encompasses three critical steps:
siRNA Generation: Initially, small interfering RNAs (siRNAs) are produced through the action of RNA polymerase IV (Pol IV).
lincRNA Production: Subsequently, long intergenic noncoding RNAs (lincRNAs) are generated via the activity of RNA polymerase V (Pol V).
Complex Formation and Methylation Establishment: Finally, the siRNAs associate with Argonaute (AGO) proteins to form complexes that pair complementarily with lincRNAs. These complexes then recruit Domain Rearranged Methyltransferase 2 (DRM2) to the target sites, thereby establishing DNA methylation.
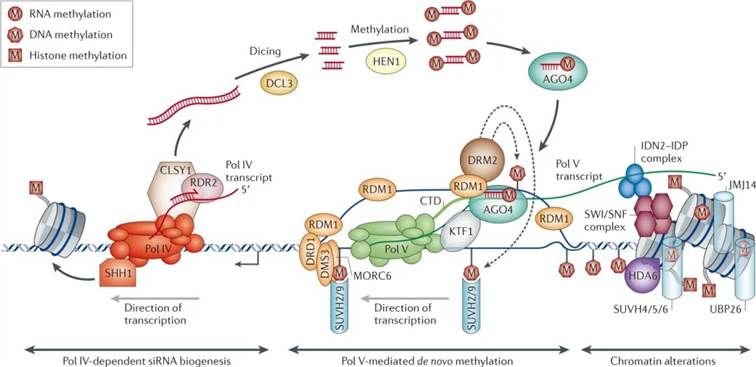 Figure 2: Model of RNA-directed DNA Methylation (RdDM) in Plants (Matzke et al., 2014). This model delineates the involvement of RNA Polymerase IV (Pol IV) in the synthesis of single-stranded RNA (ssRNA) by targeting regions enriched in transposable elements and repetitive sequences. Subsequently, RNA-dependent RNA polymerase 2 (RDR2) utilizes the ssRNA as a template to synthesize double-stranded RNA (dsRNA). The Dicer-like 3 (DCL3) enzyme then cleaves the dsRNA into 24-nucleotide small interfering RNAs (siRNAs).
Figure 2: Model of RNA-directed DNA Methylation (RdDM) in Plants (Matzke et al., 2014). This model delineates the involvement of RNA Polymerase IV (Pol IV) in the synthesis of single-stranded RNA (ssRNA) by targeting regions enriched in transposable elements and repetitive sequences. Subsequently, RNA-dependent RNA polymerase 2 (RDR2) utilizes the ssRNA as a template to synthesize double-stranded RNA (dsRNA). The Dicer-like 3 (DCL3) enzyme then cleaves the dsRNA into 24-nucleotide small interfering RNAs (siRNAs).
The maintenance methylation refers to the biochemical process that converts hemimethylated sequences, which arise post-replication, into fully methylated sequences. In plants, the maintenance of DNA methylation is closely dependent on the type of cytosine methylation. Based on the sequence context, plant DNA methylation can be categorized into three types: CG, CHG, and CHH (where H represents A, T, or C). CG and CHG methylation are symmetric, while CHH methylation is asymmetric, as classified by Law and Jacobsen in 2010. Notably, CG methylation is the predominant form of methylation in the model organism Arabidopsis thaliana, as demonstrated by Cokus et al. in 2008.
Different types of methylation are catalyzed by specific DNA methyltransferases. CG methylation maintenance is mediated by DNA Methyltransferase 1 (MET1), the plant ortholog of mammalian DNA cytosine-5 methyltransferase 1 (DNMT1). MET1 recognizes hemimethylated CG dinucleotides formed during DNA replication and catalyzes the methylation of the unmethylated cytosine on the nascent strand. CHG methylation maintenance is facilitated by Chromomethylase 2 (CMT2) or Chromomethylase 3 (CMT3). The maintenance of CHH methylation involves the activity of Domain Rearranged Methyltransferase 2 (DRM2) or CMT2. These DNA methyltransferases play crucial roles in maintaining plant DNA methylation.
Demethylation refers to the biochemical process by which methyl groups are excised from DNA sequences by demethylase enzymes, predominantly occurring at 5-methylcytosine (5-mC) residues. This phenomenon can be classified into passive and active pathways of demethylation.
Passive demethylation transpires during DNA replication when newly synthesized DNA strands are inadequately targeted by DNA methyltransferases, resulting in the progressive reduction of methylation levels over successive cell divisions. This diminution arises due to the failure of DNA methyltransferases to re-establish methylation marks on the nascent DNA strand.
Conversely, active demethylation entails the direct enzymatic conversion of 5-mC to unmodified cytosine. This mechanism is orchestrated by specific demethylase enzymes that catalyze the removal of the methyl moiety, thereby reinstating the original cytosine residue. The involvement of demethylases in active demethylation underscores the intricacy of epigenetic regulation and its pivotal role in cellular processes.
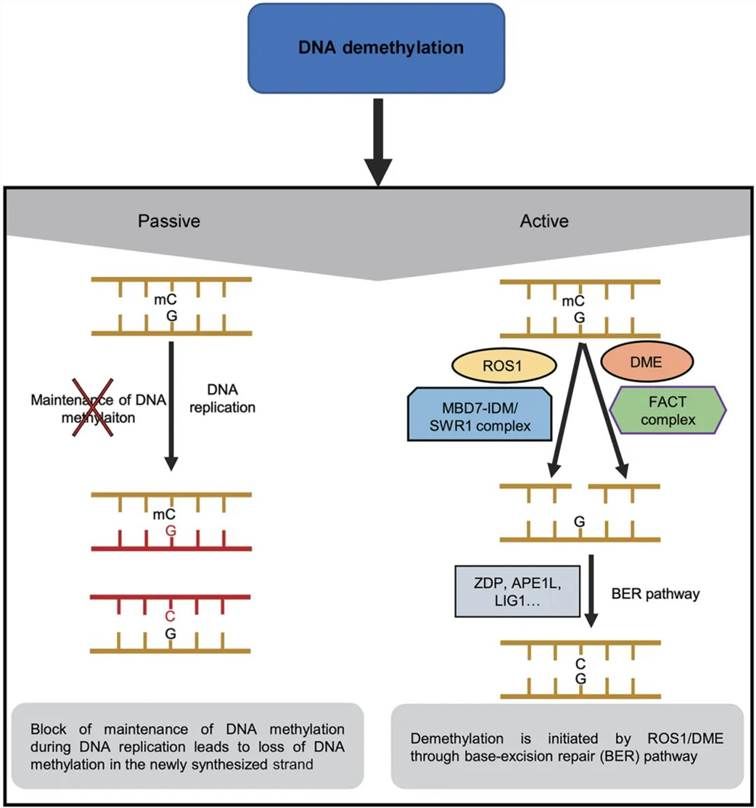 Figure 3 illustrates the mechanisms of DNA demethylation (Liu et al., 2020). Passive DNA demethylation can occur during DNA replication, resulting in a reduction of DNA methylation levels in newly synthesized DNA. Active DNA demethylation is mediated by the plant-specific ROS1/DME enzyme family. ROS1 and DME are recruited to target sites via the MBD7-IDM or SWR1 complex and the Facilitates Chromatin Transactions (FACT) complex, respectively. Upon recognition of methylated cytosine by ROS1 or DME, the N-glycosidic bond of the base is cleaved, creating an abasic site. This site is subsequently repaired by the insertion of an unmethylated cytosine, thereby completing the demethylation process at that locus.
Figure 3 illustrates the mechanisms of DNA demethylation (Liu et al., 2020). Passive DNA demethylation can occur during DNA replication, resulting in a reduction of DNA methylation levels in newly synthesized DNA. Active DNA demethylation is mediated by the plant-specific ROS1/DME enzyme family. ROS1 and DME are recruited to target sites via the MBD7-IDM or SWR1 complex and the Facilitates Chromatin Transactions (FACT) complex, respectively. Upon recognition of methylated cytosine by ROS1 or DME, the N-glycosidic bond of the base is cleaved, creating an abasic site. This site is subsequently repaired by the insertion of an unmethylated cytosine, thereby completing the demethylation process at that locus.
DNA methylation, a pivotal biological process, critically regulates gene expression, maintains genomic stability, and modulates plant growth, development, and responses to environmental stress. To enhance comprehension of DNA methylation's functions in these contexts, pertinent literature has been summarized by CD Genomics for reference. Importantly, research on DNA methylation's functions is extensive, with numerous unexplored aspects. Readers interested in this field are encouraged to consult additional literature for a comprehensive understanding.
DNA methylation participates in the regulation of fleshy fruit ripening. Knockout of the demethylase gene SlDML2 in tomato leads to elevated genome-wide methylation levels, suppressing the expression of ripening-related genes and resulting in impaired fruit ripening (Liu et al., 2015). In strawberry, overall DNA methylation levels decrease during fruit ripening (Cheng et al., 2018). However, during the development and ripening of sweet orange fruits, genome-wide DNA methylation levels increase (Huang et al., 2019).
DNA methylation is involved in regulating plant pigmentation. The promoter regions of anthocyanin biosynthesis-related gene MhMYB10 in crabapple are highly methylated, leading to reduced expression of MhMYB10 and consequently reduced accumulation of anthocyanins (Han et al., 2020). Methylation of the promoters of PrANS and PrF3H in peony leads to decreased expression in non-spotted areas of petals, thereby affecting the coloration of peony petals (Zhu et al., 2023).
In rice (Oryza sativa), iron deficiency induces the formation of hypermethylated regions predominantly of the CHH type near genes. Additionally, significant differential methylation occurs in the core transcription factors of the iron signaling pathway, OsbHLH156 and IRO2, under iron-deficient conditions (Sun et al., 2021). Under salt stress, overexpression of SlSAMS1 enhances the methylation levels of the SlGI gene body, thereby increasing the expression of SlGI and subsequently enhancing the salt tolerance of tomato (Solanum lycopersicum) plants (Chen et al., 2023).
Currently, several techniques are effective for detecting genome-wide DNA methylation in plants, including BS-seq, reduced representation bisulfite sequencing (RRBS), methylated DNA immunoprecipitation (MeDIP) sequencing, and methylation-sensitive amplified polymorphism (MSAP) technology. This article focuses on the gold standard for DNA methylation research: BS-seq.
Service you may intersted in
Learn more
BS-seq operates by treating DNA sequences with bisulfite, which converts unmethylated cytosine (C) into uracil (U), while methylated cytosines, including 5mC and 5-hydroxymethylcytosine (5hmC), remain unchanged. This treatment allows for the differentiation between methylated and unmethylated cytosines in the genome.
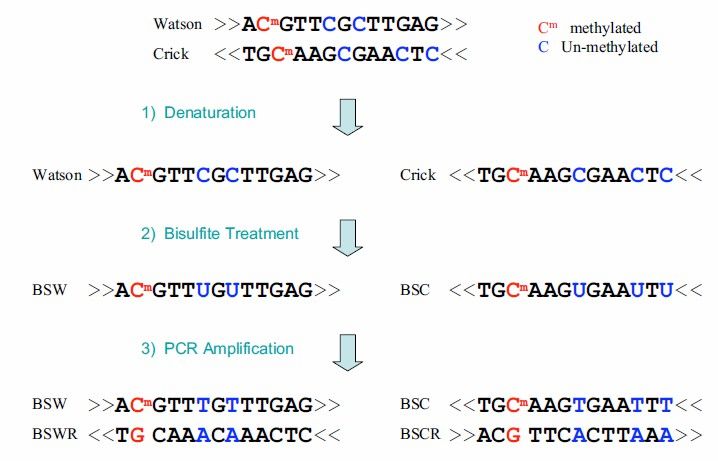 Figure 4. BS-seq technical principle. The blue nucleotides represent unmethylated C that have been converted to Uby bisulfite treatment, while the red nucleotides represent 5mC that is resistant to this conversion.
Figure 4. BS-seq technical principle. The blue nucleotides represent unmethylated C that have been converted to Uby bisulfite treatment, while the red nucleotides represent 5mC that is resistant to this conversion.
Overall, the sample is treated with bisulfite, converting unmethylated cytosines in the genome into uracils. This is followed by PCR amplification and high-throughput sequencing, enabling the generation of a single-base resolution DNA methylation map across the entire genome.
Several challenges arise when comparing BS-seq with conventional transcriptomics. First, unmethylated C are converted to T, leading to a relative decrease in cytosine and an increase in ATG sequences within the genome, which is not reflective of the true biological state. Second, standard reference genomes are used for alignment, resulting in difficulties as these converted reads may not match the corresponding loci in the reference genome accurately. Additionally, BS-seq generates four different DNA strands post-bisulfite treatment and PCR amplification, in contrast to the two complementary strands typically analyzed in transcriptomics.
BS-seq presents several notable advantages for the analysis of DNA methylation. It facilitates single-base resolution detection of both CpG and non-CpG methylation across the entire genome, enabling detailed epigenetic profiling. Additionally, BS-seq provides dense coverage, including regions of low complexity and repetitive sequences, which allows for comprehensive detection of 5mC.
Despite these advantages, BS-seq exhibits several limitations. The process of bisulfite treatment, which converts unmethylated cytosines to thymines, reduces sequence complexity, thus complicating alignment and mapping procedures. Furthermore, incomplete conversion during bisulfite treatment can result in inaccurate methylation calls. Additionally, BS-seq does not distinguish between 5mC and 5hmC, thereby limiting its capacity to fully characterize methylation states.
The application of BS-seq technology, which facilitates comprehensive analysis of DNA methylation levels and their alterations, is of paramount importance in molecular biology. This methodology effectively detects variances in DNA methylation between different samples, thereby providing critical information for advanced research. Notably, BS-seq technology plays an essential role in elucidating differences in DNA methylation at various developmental stages, contributing significantly to the understanding of epigenetic regulatory mechanisms throughout the developmental process.
Furthermore, considering the potential impact of environmental factors on DNA methylation, the utilization of BS-seq technology has elucidated how environmental influences modify gene expression via methylation, subsequently affecting plant growth and development. This insight is crucial for revealing the intricate relationship between environmental conditions and plant developmental processes.
It is noteworthy that BS-seqtechnology also permits comparative analysis of DNA methylation differences between mutants and wild-type organisms, thereby uncovering the effects of genetic mutations on methylation patterns and gene expression regulation. This capability is of substantial importance for understanding the biological consequences of gene mutations and their regulatory mechanisms.
To further comprehend the application of BS-seq technology in plant research, an in-depth discussion and analysis of three selected publications will be presented. These studies will provide a wealth of case studies and experimental data, enhancing the understanding of the significance and future prospects of BS-seq technology in the field of plant research.
In February 2024, a research paper titled "Hypermethylation in the promoter regions of flavonoid pathway genes is associated with skin color fading during 'Daihong' apple fruit development" was published in the Horticulture Research journal. The study focused on elucidating the mechanism of DNA methylation involvement in the skin color fading of 'Daihong' (DH) apples, a red-fleshed cultivar, through genetic background analysis, metabolomics, transcriptomics, methylomics, and molecular biology techniques.
Observations were conducted on the phenotypic characteristics and flavonoid content of DH fruits at seven developmental stages. It was noted that during the early stages of fruit development, the skin exhibited a deep red color with abundant flavonoid content. However, as the fruit matured, the skin color gradually lightened, with mature DH apples primarily displaying a green hue (Figure 5).
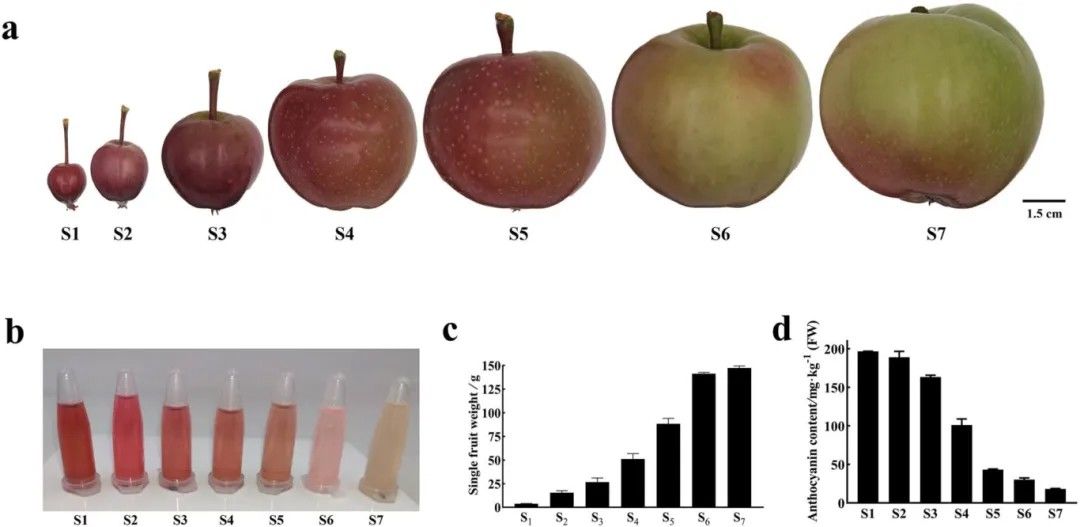 Figure 5.Phenotypic Characteristics and Flavonoid Content of DH Fruits Across Seven Developmental Stages (Xu et al., 2024).
Figure 5.Phenotypic Characteristics and Flavonoid Content of DH Fruits Across Seven Developmental Stages (Xu et al., 2024).
(a) Skin color of fruits at stages S1-S7. S1-S7 denote fruits at 30, 45, 55, 71, 90, 110, and 131 days after flowering, respectively. (b) Flavonoid extraction from DH fruit skin. (c) Individual fruit weight. (d) Determination of DH fruit skin flavonoid content using pH differential method.
To explore the DNA methylation landscape during DH fruit development, the fruit peel at developmental stages S1, S4, and S7 underwent BS-seq analysis. Results revealed a decrease in relative methylation levels of CG and CHG types, while an increase was observed in CHH type methylation as the fruit matured (Figure 6a). Overall, methylation levels across the entire genome increased during fruit development (Figure 6b). Furthermore, in all three methylation types (CG, CHG, and CHH), methylation levels in the 2 kb regions upstream or downstream of gene bodies and transposable elements (TEs) gradually increased from S1 to S7 (Figure 6c, d).
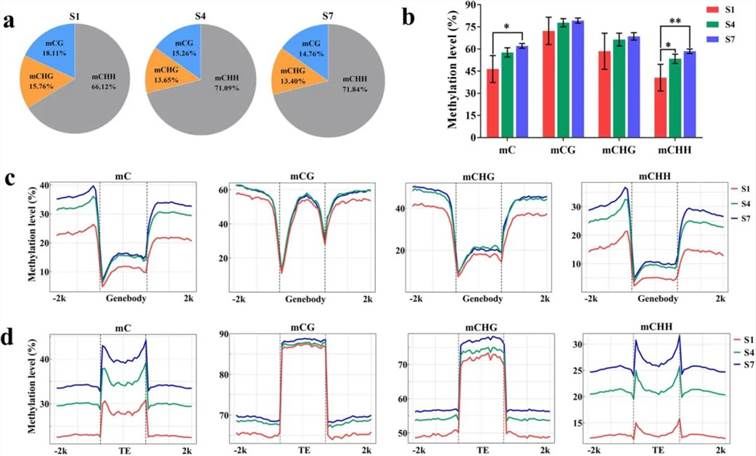 Figure 6. DNA Methylation Analysis of DH Fruit Peel (Xu et al., 2024).
Figure 6. DNA Methylation Analysis of DH Fruit Peel (Xu et al., 2024).
(a) Relative proportions of mC, mCG, mCHG, and mCHH at developmental stages S1, S4, and S7.
(b) DNA methylation levels of CG, CHG, and CHH at stages S1, S4, and S7.
(c) DNA methylation levels in the 2 kb regions upstream and downstream of genes at stages S1, S4, and S7.
(d) DNA methylation levels in the 2 kb regions upstream and downstream of transposable elements (TEs) at stages S1, S4, and S7.
Due to the influence of DNA methylation in promoter regions on gene expression, the authors conducted a correlation analysis between the methylation levels and transcriptional levels of key genes involved in anthocyanin biosynthesis and degradation pathways. It was observed that the methylation levels within the promoter regions of structural genes MdCHS, MdCHI, MdF3H, MdANS, MdUFGT, MdLAR, and MdANR exhibited an increasing trend from stages S1 to S7, while their transcriptional levels showed a decreasing trend (Figure 7). Similarly, the methylation and transcriptional levels within the promoter regions of regulatory genes MdbHLH3, MdMYB10, MdMYB16, MdMYB28, and MdMYB111 displayed an increasing trend from stages S1 to S7 (Figure 7).
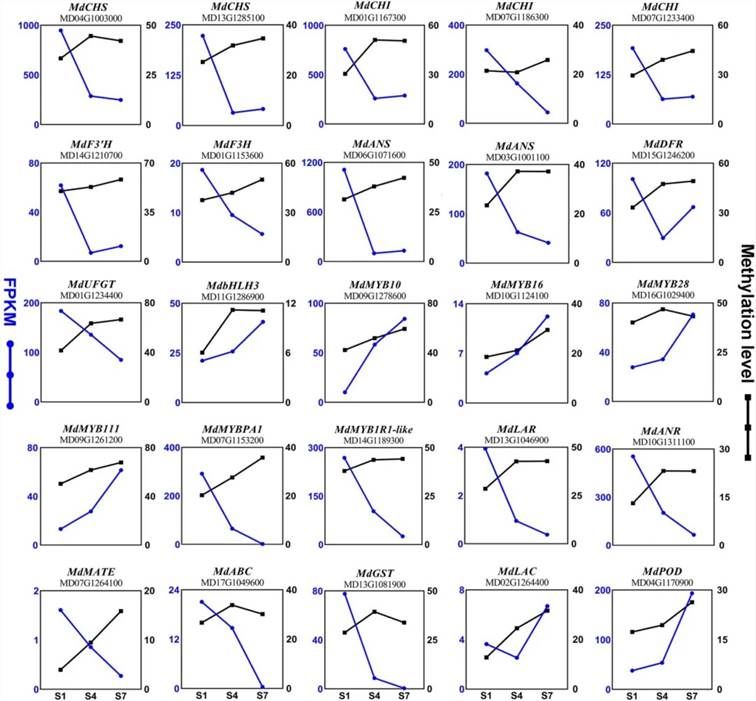 Figure 7. DNA Methylation Levels and Expression Levels of Genes Involved in Anthocyanin Accumulation (Xu et al., 2024). The blue line represents transcription levels (FPKM), while the black line represents methylation levels (%).
Figure 7. DNA Methylation Levels and Expression Levels of Genes Involved in Anthocyanin Accumulation (Xu et al., 2024). The blue line represents transcription levels (FPKM), while the black line represents methylation levels (%).
In October 2022, a research paper titled "Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis)" was published in the Plant Biology journal. Through comprehensive analysis of physiological traits, gene expression profiles, and whole-genome DNA methylation patterns of strawberries subjected to drought stress at four different time points, the authors elucidated the regulatory network underlying drought response in strawberries.
Observations were made on the morphological changes of strawberries at different time points under drought stress, alongside measurements of proline (Pro), malondialdehyde (MDA), superoxide dismutase (SOD), peroxidase (POD) levels, and relative electrical conductivity (REC). It was noted that significant variations in both morphological traits and physiological indicators occurred at the T8 time point (Figure 8).
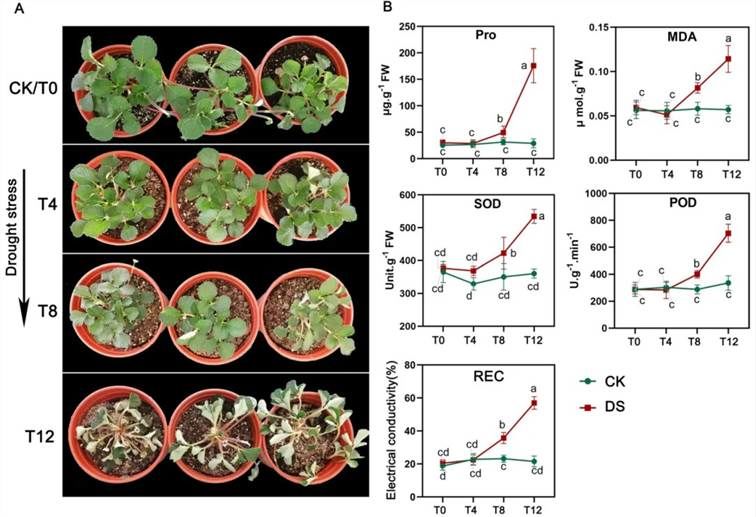 Figure 8. Analysis of Physiological Traits in Strawberry under Drought Stress (Cao et al., 2022).
Figure 8. Analysis of Physiological Traits in Strawberry under Drought Stress (Cao et al., 2022).
(A) Morphological changes in strawberry under drought stress. (B) Measurements of proline (Pro), malondialdehyde (MDA), superoxide dismutase (SOD), peroxidase (POD), and relative electrical conductivity (REC) at four time points during drought treatment.
To investigate the regulatory mechanisms of DNA methylation under drought stress, a BS-seq analysis was conducted. The results indicated that the levels of DNA methylation in the CG, CHG, and CHH contexts were 47.69%, 30.87%, and 10.56%, respectively (Figure 9A). The percentages of mCG, mCHG, and mCHH within the total DNA methylation sites exhibited dynamic changes at various drought stress time points, with mCHH not only comprising the highest proportion but also displaying the greatest variability (Figure 9B). During drought stress, strawberry (Fragaria × ananassa) promoters and repetitive sequences exhibited significant alterations in CHH methylation levels (Figure 9C). Given that promoter and genic methylation differentially influence gene expression, differentially methylated genes (DMGs) were categorized into promoter DMGs and genic DMGs. The results demonstrated that promoter DMGs were more prevalent than genic DMGs across all drought stress time points (Figure 9D). Among promoter DMGs, 317 hypomethylated (hypo) genes and 21 hypermethylated (hyper) genes were identified as common across all time points (Figure 9E). This dynamic methylation pattern suggests that these genes might directly or indirectly participate in the regulatory response to drought stress.
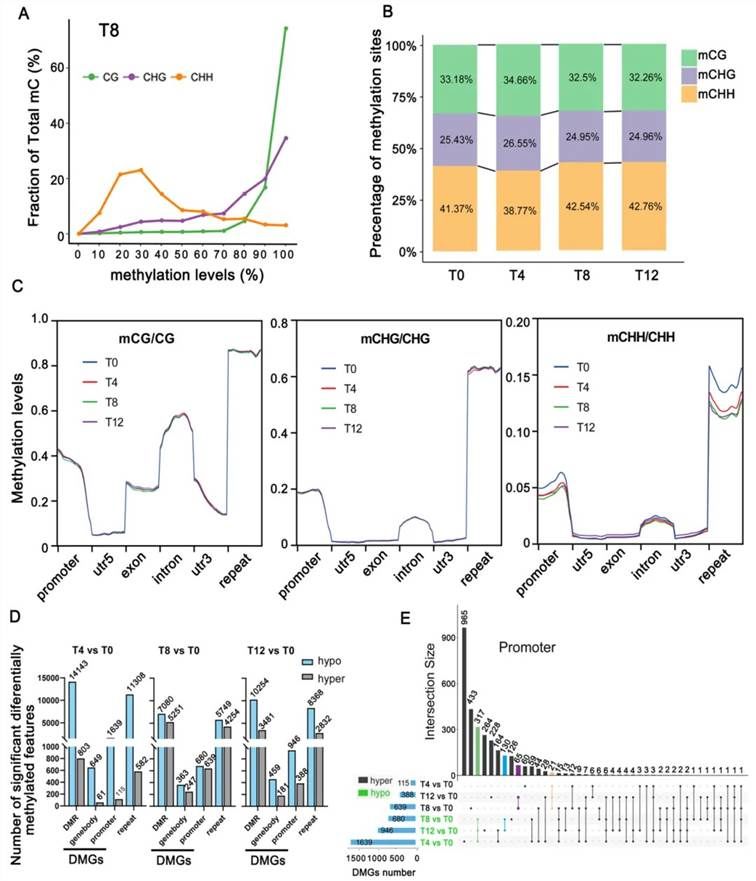 Figure 9 illustrates the changes in DNA methylation levels at four time points under drought stress (Cao et al., 2022). (A) Displays the alterations in CG, CHG, and CHH methylation levels at time point T8. (B) Represents the relative proportions of CG, CHG, and CHH methylation at different time points of drought stress. (C) Depicts the dynamic changes in DNA methylation, encompassing genomic compartments and repetitive sequences. (D) Shows the quantity of DMRs and DMGs at various time points of drought stress. (E) Presents the Upset-Venn diagram of promoter DMGs.
Figure 9 illustrates the changes in DNA methylation levels at four time points under drought stress (Cao et al., 2022). (A) Displays the alterations in CG, CHG, and CHH methylation levels at time point T8. (B) Represents the relative proportions of CG, CHG, and CHH methylation at different time points of drought stress. (C) Depicts the dynamic changes in DNA methylation, encompassing genomic compartments and repetitive sequences. (D) Shows the quantity of DMRs and DMGs at various time points of drought stress. (E) Presents the Upset-Venn diagram of promoter DMGs.
In January 2023, the journal Plant Physiology published a research paper titled "Factor of DNA methylation 1 affects woodland strawberry plant stature and organ size via DNA methylation." Through BS-seq and transcriptome analysis, the authors revealed the role of DNA methylation factor 1 (FDM1) in woodland strawberry (Fragaria vesca), demonstrating its influence on leaf, flower, and fruit size by modulating DNA methylation. This study highlighted the crucial regulatory role of RdDM in the growth and development of strawberries.
Utilizing CRISPR/Cas9 technology, the authors generated three knockout lines of FveFDM1, termed fvefdm1 (L1-L3). Comparative analysis with the wild type (WT) revealed that fvefdm1 exhibited reduced leaf size (Figure 10A-E). Additionally, the number of epidermal cells in proximal leaves of fvefdm1 significantly decreased, accompanied by an increase in cell size (Figure 10F, G). Moreover, the expression levels of FveFDM1 in young leaves of fvefdm1 were notably reduced (Figure 10H).
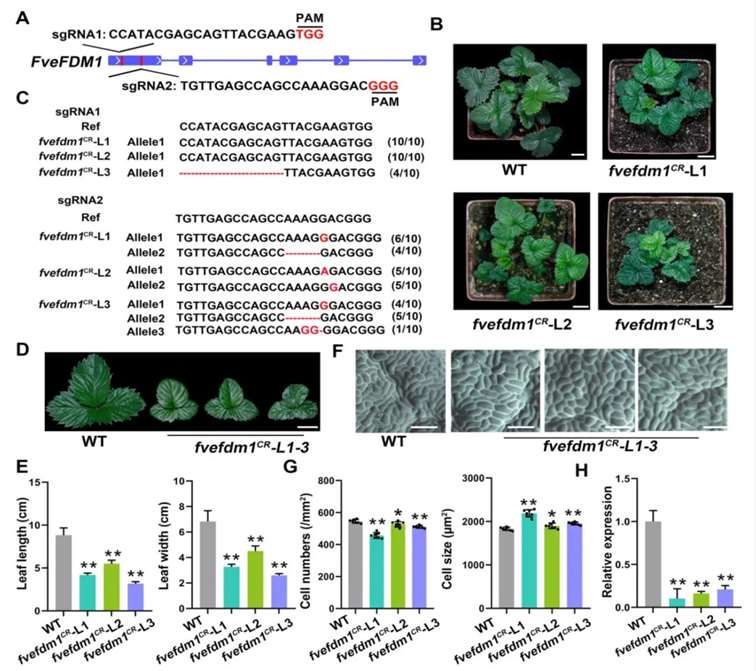 Figure 10 Analysis of fvefdm1 Mutants and Phenotypic Observation Utilizing CRISPR/Cas9 Technology (Zheng et al., 2023). (A) Schematic representation of the sgRNA target site within FveFDM1; (B) Phenotypic comparison between WT and three T0 fvefdm1 mutants (L1-L3); (C) Mutation analysis of the T0 fvefdm1 mutants (L1-L3); (D) Comparative analysis of mature leaves from fvefdm1 (L1-L3) and WT; (E) Leaf size comparison between fvefdm1 (L1-L3) and WT; (F) Analysis of adaxial epidermal cells in leaves of fvefdm1 (L1-L3) and WT; (G) Quantitative assessment of cell number and cell size in the adaxial epidermis of leaves from fvefdm1 (L1-L3) and WT; (H) Expression levels of FveFDM1 in young leaves of fvefdm1 (L1-L3) compared to WT.
Figure 10 Analysis of fvefdm1 Mutants and Phenotypic Observation Utilizing CRISPR/Cas9 Technology (Zheng et al., 2023). (A) Schematic representation of the sgRNA target site within FveFDM1; (B) Phenotypic comparison between WT and three T0 fvefdm1 mutants (L1-L3); (C) Mutation analysis of the T0 fvefdm1 mutants (L1-L3); (D) Comparative analysis of mature leaves from fvefdm1 (L1-L3) and WT; (E) Leaf size comparison between fvefdm1 (L1-L3) and WT; (F) Analysis of adaxial epidermal cells in leaves of fvefdm1 (L1-L3) and WT; (G) Quantitative assessment of cell number and cell size in the adaxial epidermis of leaves from fvefdm1 (L1-L3) and WT; (H) Expression levels of FveFDM1 in young leaves of fvefdm1 (L1-L3) compared to WT.
To investigate the molecular function of FveFDM1, the DNA methylation levels in WT and fvefdm1 were examined using BS-seq. It was found that, compared to WT, the average methylation levels of CG and CHG types in all gene bodies and transposable elements (TEs) of fvefdm1 remained largely unchanged, while the average methylation level of the CHH type significantly decreased (Figure 11A). Subsequently, differential methylation regions (DMRs) were identified between fvefdm1 and WT, yielding a total of 16,831 CG DMRs, 46,264 CHG DMRs, and 74,749 CHH DMRs, with the majority being hypo DMRs (Figure 11B). Moreover, hypo DMRs exhibited a significant decrease in DNA methylation levels in fvefdm1 (Figure 11C). Hypo DMRs of all three methylation types were predominantly enriched either in gene body regions only or in regions covering both gene bodies and TEs (Figure 11D). Specifically, hypo DMRs in all three scenarios were relatively abundant in gene promoters (Figure 11E). These findings suggest that FveFDM1 regulates DNA methylation at positions most likely to influence gene expression.
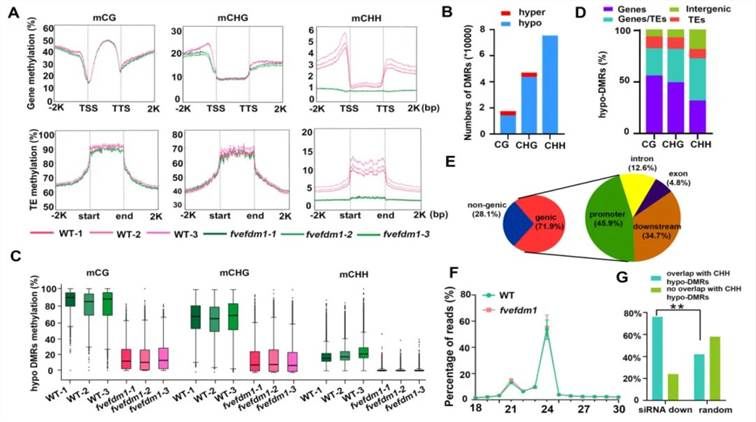 Figure 11 illustrates the DNA methylation levels and siRNA abundance in fvefdm1 (Zheng et al., 2023). (A) Average methylation levels of CG, CHG, and CHH in all genes and transposable elements (TEs) in both WT and fvefdm1, including 2 kb upstream of the transcription start site (TSS) and 2 kb downstream of the transcription termination site (TTS). (B) Number of hyper-DMRs and hypo-DMRs in CG, CHG, and CHH relative to WT in fvefdm1. (C) Box plots showing the methylation levels of CG, CHG, and CHH in hypo-DMRs in WT and fvefdm1. (D) Genomic distribution of hypo-DMRs in CG, CHG, and CHH. (E) Distribution of CHH hypo-DMRs in different genomic regions. "Promoter" indicates 2 kb upstream of TSS, and "Downstream" indicates 2 kb downstream of TTS. (F) Size distribution of siRNAs aligned to the genome in WT and fvefdm1. (G) Percentage of downregulated (n=23395) or random (n=23395) siRNA clusters overlapping or not overlapping with CHH-hypo-DMRs.
Figure 11 illustrates the DNA methylation levels and siRNA abundance in fvefdm1 (Zheng et al., 2023). (A) Average methylation levels of CG, CHG, and CHH in all genes and transposable elements (TEs) in both WT and fvefdm1, including 2 kb upstream of the transcription start site (TSS) and 2 kb downstream of the transcription termination site (TTS). (B) Number of hyper-DMRs and hypo-DMRs in CG, CHG, and CHH relative to WT in fvefdm1. (C) Box plots showing the methylation levels of CG, CHG, and CHH in hypo-DMRs in WT and fvefdm1. (D) Genomic distribution of hypo-DMRs in CG, CHG, and CHH. (E) Distribution of CHH hypo-DMRs in different genomic regions. "Promoter" indicates 2 kb upstream of TSS, and "Downstream" indicates 2 kb downstream of TTS. (F) Size distribution of siRNAs aligned to the genome in WT and fvefdm1. (G) Percentage of downregulated (n=23395) or random (n=23395) siRNA clusters overlapping or not overlapping with CHH-hypo-DMRs.
Furthermore, it was observed that the expression of FveFDM1 and FveIDN2 proteins can be induced by GA. The protein complex formed by FveFDM2 and FveIDN2 directly interacts with AGO4 siRNA, thus influencing the DNA methylation level at target sites. Consequently, downstream regulatory genes are either promoted or inhibited by DNA methylation, leading to alterations in organ size by interfering with cell division.
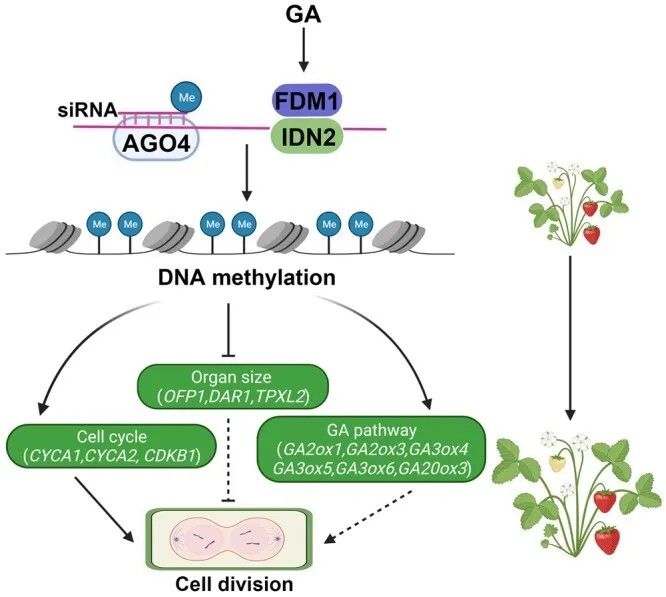 Figure 12 Functional model of the FveFDM1 gene in strawberry (Zheng et al., 2023).
Figure 12 Functional model of the FveFDM1 gene in strawberry (Zheng et al., 2023).
In this scholarly article, CD Genomics systematically elucidates the fundamental patterns of DNA methylation in plants. The exposition delineates the biological process of DNA methylation as a dynamic interplay among de novo methylation, maintenance methylation, and demethylation mechanisms. Furthermore, it briefly outlines the pivotal roles of DNA methylation in plant growth, development, and response to environmental stresses, underscoring its significance in plant life.
Lastly, the article focuses on the BS-seq technique, widely acknowledged as the "gold standard" for DNA methylation detection, and thoroughly explores its applications across various domains. It is anticipated that this article will offer insights and perspectives to researchers in related fields, guiding their exploration and inquiry.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.