







We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In recent years, RNA modification research has gradually become an important area of epitranscriptomics, in which N4-acetylcytidine (ac4C), an emerging RNA chemical modification, has attracted much attention because of its role in regulating mRNA stability, translation efficiency, and gene expression. Early studies have found that ac4C modifications are predominantly distributed in eukaryotic tRNAs and rRNAs and improve translation accuracy by enhancing the thermal stability of base pairing; In contrast, recent studies have shown that ac4C is widely distributed in the human transcriptome, particularly within the coding sequence (CDS), and is able to significantly regulate target gene expression by improving mRNA stability and translational efficiency. This finding provides new perspectives for understanding the role of RNA modifications in biological processes and diseases.
To systematically analyze the distribution of ac4C modifications and their functions, Acetylated RNA ImmunoPrecipitation Sequencing (acRIP-seq) technology was developed. This technology combines RNA immunoprecipitation and high-throughput sequencing to efficiently and accurately identify transcriptome-wide ac4C modification sites and reveal their biological significance through bioinformatics analysis. In the next sections, we will delve into the principles of acRIP-seq technology and experimental workflow.
Service you may intersted in
If you want to learn more about the acRIP-seq, please refer to:
As a high-throughput sequencing technology dedicated to the detection of ac4C modifications, the core of acRIP-seq is the enrichment of ac4C-modified RNA fragments by specific antibodies and the combination of deep sequencing and bioinformatics tools to achieve transcriptome-wide modification localization.The ac4C modification is catalyzed by RNA modifying enzymes such as NAT10 and is distributed in tRNAs, rRNAs, and mRNAs, which not only enhances base-pair stability, but also mRNA stability and translational efficiency, and plays an important role in gene regulation and diseases such as cancer.
Generation of an epitranscriptome by chemical modification of mRNAs has emerged as a novel mechanism for post-transcriptional gene regulation, and while most mRNA modifications are methylation events, a single acetylated ribonucleoside has been described in eukaryotes, occurring at the N4 position of the cytidine. The production of ac4C is dependent on the catalytic action of the NAT10 enzyme or its homologs and exhibits a unique nonredundant activity that highlights its functional specificity in RNA modification.
Studies have shown that ac4C modifications are significantly enriched in the coding sequence (CDS) region of mRNAs, and their functions are closely related to mRNA stability and translation efficiency. NAT10 enzyme deficiency results in the loss of modification of specific ac4C targets, which triggers a significant reduction in mRNA expression levels. In contrast, when ac4C modifications were present in the CDS, mRNA stability was enhanced and translational efficiency was significantly increased.
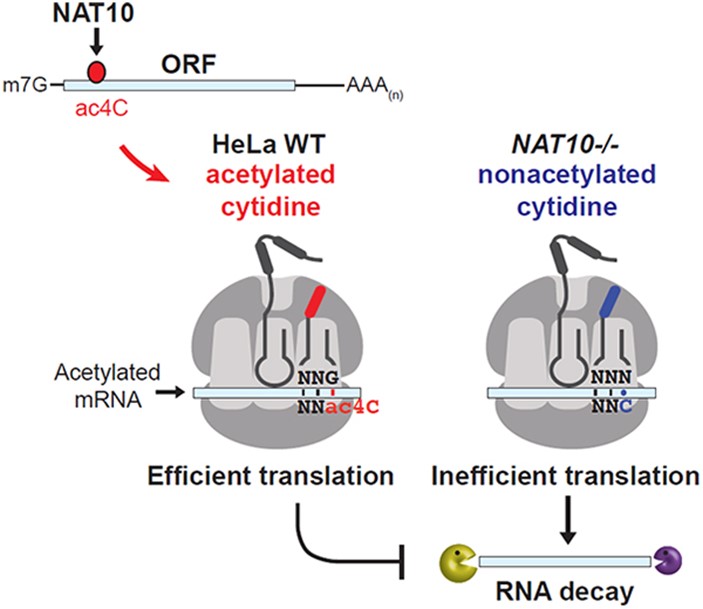 Graphical Abstract of Facilitation of translation efficiency by ac4C (Arango et al., 2018)
Graphical Abstract of Facilitation of translation efficiency by ac4C (Arango et al., 2018)
NAT10 enzymes play a key role in N4-acetylcytidine (ac4C) modification in mRNAs. This modification is widely distributed in the coding sequence (CDS) and reveals its biological function during translation. The images clearly demonstrate the promotional effect of ac4C modification on translational efficiency: in wild-type HeLa cells, ac4C improves the translational efficiency of mRNAs by enhancing the efficiency of codon-anti-codon pairing at the ribosome. In contrast, in NAT10 knockout cells, the absence of the ac4C modification resulted in a significant decrease in translation efficiency, reduced RNA stability, and an increased risk of RNA degradation.
Molecularly, ac4C works by immobilizing cytosine in the "proximal" conformation, allowing it to form stable Watson-Crick base pairs, especially at codon swing sites.This ability to enhance pairing is critical to the accuracy and efficiency of translation. After the cytosine at the swing site (codon third) is acetylated, more efficient binding is achieved with the anticodon in the tRNA, reducing translation errors. It was further shown that ac4C modification provides a significant advantage for codons with multiple oscillatory site selectivity, especially codon 1 and 2 sites containing A/U combinations. This phenomenon is similar to the mechanism by which tRNA acetylation enhances translation accuracy in bacteria.
Overall, ac4C not only enhances mRNA stability and translational efficiency, but may also have a profound impact on the regulation of gene expression by affecting multiple levels of ribosome decoding, translation initiation, and RNA stability.
The ac4C acetylation modification was first reported in rRNAs and tRNAs, and the modification is regulated by the NAT10 single enzyme system, which possesses both acetylation catalytic function and RNA-binding activity; other catalytic enzymes and specific RBPs for ac4C have not yet been reported.
NAT10 is an important acetyltransferase that is widely present in human cells and is involved in the regulation of a variety of biological processes. As a class of dual acetyltransferases, NAT10 possesses not only protein acetylation but also RNA-binding activity. It affects mRNA stability and translation by catalyzing N4-acetylcytidine modifications, and its enzymatic activity is regulated by cell type specificity. It has been shown that NAT10 needs to interact with specific adaptor proteins when functioning in mRNAs, especially in translation initiation regions and coding sequences, and that the high-frequency region of ac4C modification is closely related to translation efficiency.
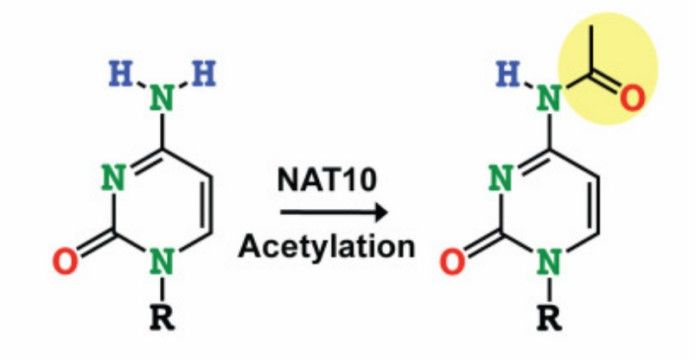 NAT10 catalyzes cytidine acetylation (Arango et al., 2019)
NAT10 catalyzes cytidine acetylation (Arango et al., 2019)
The structural features of NAT10 enable it to perform a wide range of substrate modifications, and its acetyltransferase activity is not limited to tRNAs and rRNAs, but also acts on a wide range of proteins, including alpha-microtubulin and histones. The activity of NAT10 is regulated by cellular states and transcription factors, and the RNA-binding region of NAT10 is key to the regulation of its activity. In particular, NAT10 regulates tRNA acetylation through binding to adaptor proteins such as THUMPD1, while the mechanism of action in mRNAs is not fully understood. It was noted that acetylation of NAT10 may regulate gene expression by enhancing mRNA stability and facilitating translation, especially in response to cellular stress, where the level of ac4C was significantly increased.
In addition, NAT10 plays an important role in a variety of cancers and has been recognized as a potential therapeutic target for accelerated aging and nuclear fibrinopathy-related diseases. With the deeper study of ac4C in mRNA, the role of NAT10 has become clearer and has become an important focus for studying post-transcriptional regulation.
Compared to traditional methods, acRIP-seq offers significant advantages in terms of sensitivity and specificity. The technology covers the entire transcriptome, is applicable to a wide range of sample types, and supports dynamic modification studies. Combined with other histology technologies (e.g., RNA-seq, Ribo-seq), acRIP-seq provides the ability to comprehensively resolve RNA modifications, advancing the development of epitranscriptomics in basic research and medical applications.
The core steps of an acRIP-seq experiment include cell lysis, RNA extraction, RNA fragmentation, RNA immunoprecipitation (RIP), elution and purification, library construction, and high-throughput sequencing. First, researchers obtain RNA from cells or tissues through cell lysis and RNA extraction, which ensures the purity and integrity of RNA samples and provides reliable raw materials for downstream experiments. Subsequently, RNA fragmentation allows the RNA molecule to be cut into small fragments suitable for capture, followed by RNA immunoprecipitation with specific antibodies to enrich RNA with ac4C modifications. The immunoprecipitated RNA is further purified by washing and treatment with Proteinase K, resulting in pure ac4C-modified RNA. The next step in the library construction step is by rRNA removal, reverse transcription and PCR amplification to convert the RNA into libraries that can be used for high-throughput sequencing, ensuring that sufficient target sequences are captured and analyzed for accurate sequencing.
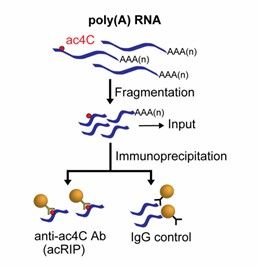 Schematics of acRIP-seq (Arango et al., 2019)
Schematics of acRIP-seq (Arango et al., 2019)
In the acRIP-seq experiment, the first step is the isolation of mRNA from total RNA, typically achieved through the enrichment of poly(A) RNA. Since mRNA molecules generally possess a poly(A) tail at their 3' end, while non-coding RNA and other types of RNA lack this feature, poly(A) RNA can be selectively captured using Dynabeads™ oligo(dT)25 magnetic beads that specifically bind to the poly(A) tail. In this process, 1.8 mg of total RNA is dissolved in 2.4 ml of nuclease-free water to achieve a concentration of 750 μg/ml. Then, 6 mg of Dynabeads® Oligo(dT)25 are resuspended and transferred to a 2.0 ml microcentrifuge tube. After separating the Dynabeads using a magnetic rack, they are washed with poly(A) RNA wash buffer once. Next, the Dynabeads are resuspended in 1.2 ml of poly(A) RNA binding buffer, and 600 μl of total RNA is added to the Dynabeads mixture. The sample is rotated at room temperature for 5 minutes to allow the mRNA to bind to the beads. Following this, the beads are separated from the supernatant using a magnetic rack, and the wash step is repeated three times to eliminate impurities. Finally, the purified poly(A) RNA is eluted using 10 mM Tris-HCl (pH 7.5) buffer. After several washes and precipitation steps, approximately 400 μl of poly(A) RNA is obtained.
RNA fragmentation is a critical step in the acRIP-seq procedure aimed at cutting mRNA into shorter fragments suitable for sequencing. For this purpose, 20 μg of poly(A) RNA is mixed with 10x RNA fragmentation buffer and divided into four 50 μl PCR tubes. The mixture is then incubated at 94oC for 5 minutes, causing RNA to break into fragments. To halt the fragmentation reaction, the tubes are immediately placed on ice. After adding RNA fragmentation termination solution, the reactions are combined and precipitated to remove impurities from the process, resulting in fragmented RNA. These RNA fragments will then be used in the subsequent acetylation process during in vitro transcription.
To generate acetylated RNA, acRIP-seq employs an in vitro transcription method to synthesize RNA modified with ac4C. In vitro transcription is typically performed using the MAXIscript T7 transcription kit, which can synthesize RNA from a DNA template that contains a T7 promoter. First, the DNA template with the T7 promoter is mixed with the transcription reaction mixture, and RNA is synthesized under the action of T7 RNA polymerase. After the transcription process, DNase I is used to eliminate any residual DNA. Then, RNA is purified using Agencourt RNAclean XP beads to remove excess reaction components, yielding purified acetylated RNA. This acetylated RNA serves as the foundation for subsequent immunoprecipitation and library construction.
In acRIP-seq, an anti-ac4C antibody is used to enrich acetylated RNA. To enable the antibody to bind to magnetic beads, it is first conjugated with Protein G Dynabeads. By incubating Protein G magnetic beads with the anti-ac4C antibody or an isotype control IgG, an antibody-bead complex is formed that specifically binds to ac4C-modified RNA. This step is crucial to ensure effective binding between the antibody and the beads, which is essential for the subsequent high-efficiency and specific immunoprecipitation.
RNA immunoprecipitation is the core step in the acRIP-seq experiment, designed to enrich target RNA that contains ac4C modifications. The fragmented RNA is incubated with magnetic beads conjugated with the anti-ac4C antibody, typically at room temperature, to allow sufficient binding between the RNA and the antibody. After incubation, the RNA-antibody-bead complexes are separated using a magnetic rack, and non-specifically bound RNA is removed through multiple washing steps. Finally, the ac4C-modified RNA is eluted from the beads using an elution buffer. The enriched RNA is then used for subsequent library construction and sequencing analysis.
The enriched RNA samples need to be converted into sequencing libraries. In acRIP-seq, NEBNext® Ultra™ II directional RNA library construction kits are used for this purpose. The process includes several critical steps: First, the enriched RNA is reverse-transcribed into complementary DNA (cDNA). Next, the cDNA undergoes end repair and adapter ligation to ensure that the cDNA ends have the necessary sequencing adapter sequences. Finally, PCR amplification is carried out to generate a sufficient amount of library fragments. After library construction, the RNA library is ready for high-throughput sequencing.
The constructed library is subjected to paired-end sequencing on the Illumina platform, typically using a 2 x 125 bp read length. High-throughput sequencing technologies like Illumina generate vast amounts of short sequence reads. These reads can then be processed to accurately identify regions of ac4C-modified RNA, providing valuable insights into the role of ac4C modifications in mRNA metabolism. Through this sequencing process, researchers can deeply analyze the characteristics of ac4C modifications and their potential functions in gene expression regulation.
acRIP-seq has proven to be a transformative technology in epitranscriptomics, offering a comprehensive method for detecting and analyzing ac4C RNA modifications. By leveraging the specificity of antibodies and high-throughput sequencing, acRIP-seq not only enhances our understanding of RNA stability and translational efficiency but also provides valuable insights into the regulation of gene expression. As research continues to evolve, this technology promises to deepen our knowledge of RNA modifications and their implications in various biological processes and diseases, including cancer.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.