Alternative Splicing
Alternative splicing is a fundamental cellular process that allows a single gene to code for multiple protein isoforms, greatly expanding the functional diversity of the proteome. Accurate characterization of alternative splicing events is essential for understanding gene regulation, tissue-specific functions, and disease mechanisms.
Traditional short-read sequencing techniques, while effective in many contexts, often fall short in accurately capturing the intricate landscape of alternative splicing due to the limitations of short read lengths and high sequencing error rates. PacBio's Iso-Seq technology has emerged as a powerful tool for comprehensive transcriptome analysis, offering high-throughput, long-read sequencing that facilitates the exploration of alternative splicing with unprecedented accuracy and depth.
Recent studies have compellingly demonstrated that over 90% of human genes undergo extensive alternative splicing, yielding a diverse array of isoforms. Remarkably, certain genes exhibit an astonishing repertoire of more than 10 isoforms. Concurrently, these dynamic splicing patterns frequently hold pivotal implications for growth, disease susceptibility, tumorigenesis, and various forms of cancer. Astonishingly, conventional short-read sequencing methods often fail to capture a substantial portion—ranging from 60% to 80%—of these intricate transcriptomic nuances. Enter the Iso-Seq investigation, built upon the ingenious PacBio technology, which offers an illuminating solution by enabling comprehensive detection of full-length cDNA molecules, substantially augmenting our capacity to unearth these elusive isoforms.
Case Study 1: Iso-Seq Reveals Transcript Diversity in Breast Cancer
Breast cancer is a heterogeneous disease characterized by a wide range of transcriptome alterations that contribute to tumor progression and patient outcomes. One of the key mechanisms underlying transcriptome diversity is alternative splicing, which generates multiple mRNA isoforms from a single gene. However, the full spectrum of isoform-level selective splicing subtypes in breast cancer remains largely unknown. Traditional short-read sequencing methods have limitations in accurately capturing full-length isoforms and identifying tumor-specific splicing events. In this case study, the researchers aimed to comprehensively identify and annotate full-length isoforms and tumor-specific splicing events in breast cancer using three generations of Iso-seq technology.
The study employed the Iso-seq technology, which is based on single-molecule long-read sequencing. This approach allows for the direct sequencing of full-length cDNAs, enabling the identification of complete isoforms and accurate detection of complex splicing events. The researchers obtained breast cancer samples from a diverse cohort of patients representing various breast cancer subtypes. They performed Iso-seq sequencing on these samples, generating high-quality long-read transcriptomic data.
To identify and annotate full-length isoforms, the researchers developed a bioinformatics pipeline that involved error correction, isoform clustering, and consensus sequence generation. They also utilized reference genome annotations to distinguish between known and novel isoforms. To identify tumor-specific splicing events, the isoform-level expression profiles of tumor samples were compared with normal tissue samples.
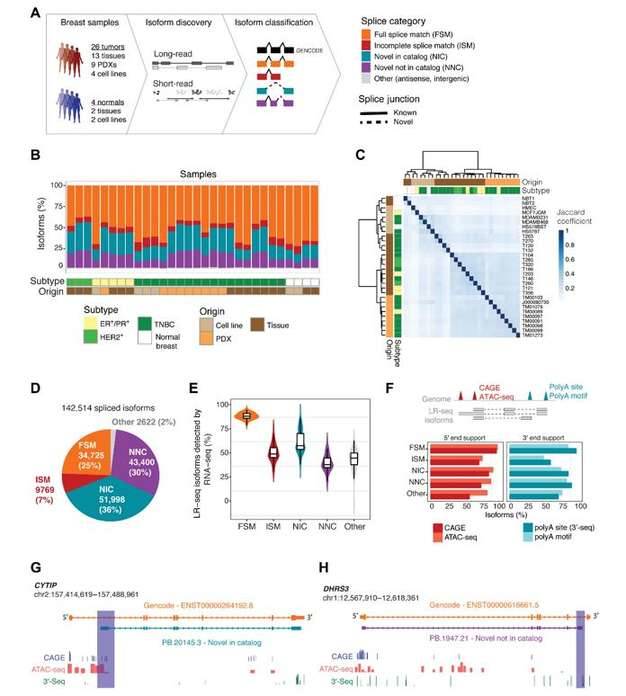 LR-seq identifies previously undetected isoforms in breast cancer. (Veiga et al., 2022)
LR-seq identifies previously undetected isoforms in breast cancer. (Veiga et al., 2022)
Remarkably, 30% of these novel isoforms exhibited alterations affecting protein-coding exons, indicating the potential functional significance of these isoforms in tumor biology. Extensive cross-validation of the identified isoforms and splicing events was performed, enhancing the robustness of the findings.
Overall, the researchers identified 3,059 breast tumor-specific splicing events that were not previously characterized. Notably, 35 of these splicing events were significantly associated with patient survival, suggesting their potential as prognostic markers. Among these genes, 21 were absent from existing databases and literature, highlighting the power of long-read sequencing in discovering novel transcriptomic features. Additionally, 10 genes were found to be enriched in specific breast cancer subtypes, providing insights into the subtype-specific splicing landscape.
Case Study 2: Iso-Seq Reveals Promoter Diversity in Different Subtypes of Gastric Cancer
Dysregulated gene expression is a defining characteristic of cancer, playing a critical role in tumor development and progression. Traditional gene expression analysis methods, often based on second-generation sequencing, have been limited by their inability to capture full-length transcripts, potentially missing important information about the true transcriptional landscape in cancer cells. To overcome this limitation, a comprehensive study was conducted to investigate the transcriptional profile of gastric cancer (GC) using a combination of Iso-seq and RNA-seq technologies. This study aimed to uncover the diversity of promoter usage and alternative splicing across different subtypes of gastric cancer.
The study focused on analyzing the full-length transcriptome of 10 gastric cancer cell lines, encompassing four distinct GC subtypes. To achieve this, the researchers employed a two-pronged approach involving Iso-seq and RNA-seq technologies.
Iso-seq Technology: Iso-seq, a state-of-the-art sequencing method, was utilized to capture full-length transcripts. This method provided longer read lengths compared to traditional sequencing, allowing for a more accurate representation of the entire transcript. This approach enabled the identification of novel transcripts that might have been missed by previous sequencing techniques.
RNA-seq Technology: In addition to Iso-seq, RNA-seq was employed to obtain quantitative gene expression data. This technology provided information about the relative abundance of different transcripts in the cell lines.
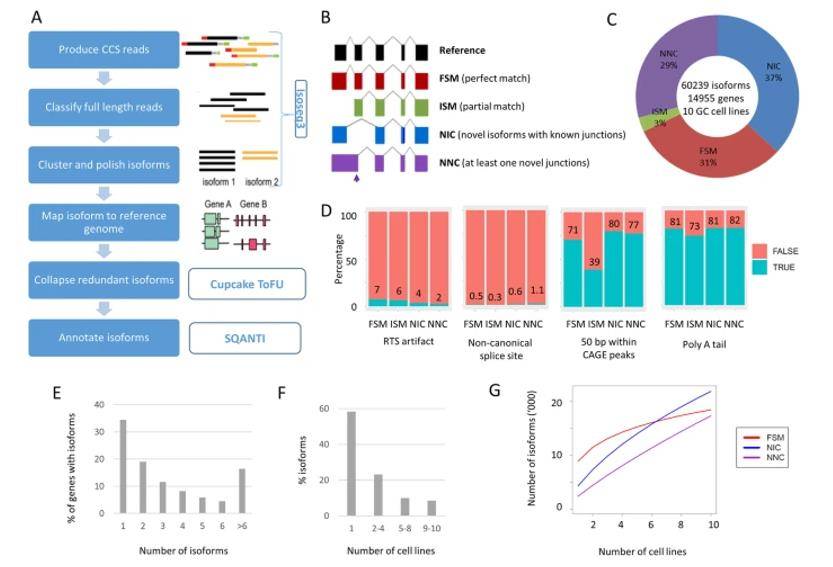 Landscape of long-read transcriptome in gastric cancer cell lines. (Huang et al., 2021)
Landscape of long-read transcriptome in gastric cancer cell lines. (Huang et al., 2021)
A significant number of non-redundant full-length transcripts were identified, with over 60% representing newly discovered transcripts. This highlights the power of lengthwise sequencing in capturing previously unannotated elements of the transcriptome. Novel transcripts were found to be subtype-specific, suggesting a potential link between these transcripts and the distinct subtypes of gastric cancer. Additionally, there appeared to be a subtype-dependent preference for alternative splicing. The newly discovered transcripts exhibited more complex structural features compared to previously known transcripts, indicating the existence of intricate regulatory mechanisms involved in these subtypes of gastric cancer. Alternative promoters were identified in approximately 25% of the genes analyzed. Importantly, many of these alternative promoters were associated with functional changes in the proteins they encoded, potentially impacting disease progression. The comprehensive dataset generated by this study has implications beyond its immediate findings. It is anticipated that the identified transcript variants will offer enhanced diagnostic potential and contribute to more accurate prognostic assessments for different subtypes of gastric cancer.
The study's results also provided a valuable resource of full-length transcriptome data, not only for further in-depth exploration of gastric cancer but also for the study of other gastrointestinal malignancies.
References:
-
Veiga, Diogo FT, et al. "A comprehensive long-read isoform analysis platform and sequencing resource for breast cancer." Science Advances 8.3 (2022): eabg6711.
- Huang, Kie Kyon, et al. "Long-read transcriptome sequencing reveals abundant promoter diversity in distinct molecular subtypes of gastric cancer." Genome biology 22 (2021): 1-24.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


