In contemporary breeding practices, it is essential to delve into the intricate genetic interactions occurring not only at the CMS and Rf loci but also at other loci that influence plant fertility, the choice between self-pollination and cross-pollination, and overall hybridization performance. This understanding is pivotal for the advancement of unimpeded cross-breeding initiatives.
This study unveiled a remarkable expansion and diversity within the RFL and mTERF gene families connected to fertility restoration. Furthermore, it shed light on the evolutionary mechanisms governing these genes, opening up new vistas for enhancing hybrid production in wheat and related varieties.
Genome-Wide Comparative Analysis of RFL Gene Families in Cereals
The inaugural genome-wide comparative investigation of the RFL (reproductive restoration) gene family in rice and barley has unveiled a remarkable degree of structural and copy number variation, both within and between species. In-depth scrutiny, including covariance and sequence conservation analyses, reveals that RFL sequences clustered together are more likely to undergo diversification compared to those dispersed outside of these clusters. This diversity within the RFL branch is driven by intricate genetic interactions between CMS genes and Rf genes, which ultimately lead to sterility.
Notably, the wheat genome harbors an unusually high abundance of RFL genes, with "Chinese Spring" alone possessing 207 such genes, a striking contrast to other angiosperms, where the count typically ranges from 20 to 30. In pursuit of these insights, researchers devised an RFL sequence capture method, akin to approaches employed for R genes. Custom-designed RFL-specific probes were utilized for sequence capture, using RFL sequences sourced from bread wheat, its subgenomic donors, and related varieties. Candidate genes responsible for the recovery of Rf1 and Rf3 were identified through comparisons between sequences found in recovered and non-recovered genotypes, with their functionalities subsequently confirmed through transgenic experiments.
Leveraging the availability of reference genome sequences for rye and wheat, researchers successfully characterized the Rfmulti multilocus (Restoration of fertility in multiple CMS systems). This endeavor led to the identification of a candidate gene for Rfmulti within the wheat genome through comparative analysis of conserved regions shared between rye and wheat genomes. The forthcoming phase involves experimental validation of this prediction.
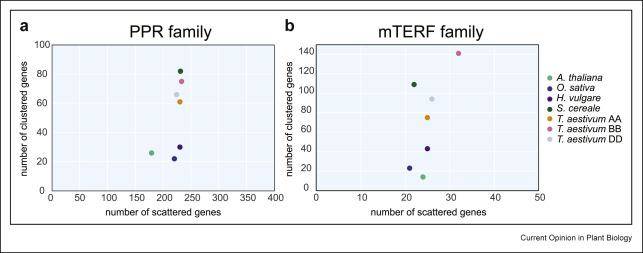 Expansions of the RFL and mTERF gene families in Triticeae and relatives. (Melonek et al., 2022)
Expansions of the RFL and mTERF gene families in Triticeae and relatives. (Melonek et al., 2022)
Discovery of RFL-type mTERF Genes in the Wheat Pan-Genome
Previous studies on rye and barley have suggested that members of the mTERF (mitochondrial transcription termination factor) family may play a crucial role in fertility restoration in cereals. These proteins are characterized by a repetitive motif spanning approximately 30-35 amino acids, known as the mTERF motif, which bears resemblance to PPR proteins. In cereals, a noteworthy similarity exists between the distribution of dispersed and clustered mTERF genes. Approximately 21-32 dispersed genes align with the 24 dispersed genes identified in Arabidopsis, many of which exhibit high conservation and likely represent homologous counterparts. In contrast, clustered mTERF genes undergo significant amplification in cereals, with over 300 in wheat and approximately 100 in rye. The clustered mTERFs exhibit an evolutionary pattern reminiscent of the RFL-PPR gene branch, characterized by high similarity among paralogs but relatively lower similarity between direct lineages. In rye, the RFL and mTERF clusters on chromosome 4R overlap with genomic regions housing the Rfp1, Rfp2, and Rfp3 recovery loci. Among these, the sequence of Rfp1 is the only one to have been cloned and characterized as an mTERF gene.
The accuracy of complex RFL and mTERF gene cluster assemblies has been significantly enhanced due to advancements in sequencing and assembly strategies tailored to cereal genomes rich in repetitive sequences. This is evident in the number of RFL genes obtained through four distinct assemblies. Refined reference genome assemblies, in conjunction with extensive pan-genomic and pan-transcriptomic datasets featuring long read-length sequences, will prove invaluable in unraveling the diversity and expansion of RFL and mTERF genes in cereals.
Mitochondrial Sequence Evolution and Its Impact on CMS Trait Maintenance
Rearrangements in the mitochondrial genome are a common occurrence in plants and often lead to the emergence of new open reading frames (ORFs), some of which contribute to male sterility. All known CMS genes exhibit mosaic structures, granting male-sterile plants an advantage over hermaphrodites in seed production by redirecting the energy allocated to pollen development toward seed production. This, in turn, promotes the transmission of the CMS mitochondrial genome through maternal inheritance. Nevertheless, the consequent proliferation of male-sterile plants creates significant selection pressure in favor of Rf genes, which act to counteract CMS. These Rf-carrying plants can then pollinate the male-sterile individuals that do not carry Rf genes.
Notably, in the context of the Ogu-CMS system in radish, a recent discovery pinpointed a single nucleotide substitution within the orf138 gene sequence as the trigger for male sterility. Intriguingly, this mutation was found within the binding site of the recovery protein Rfo and resulted in a significant reduction in Rfo's affinity for this site. Furthermore, this study unveiled a new restoration gene, Rfs, which is capable of effectively suppressing sterility induced by the mutated orf138.
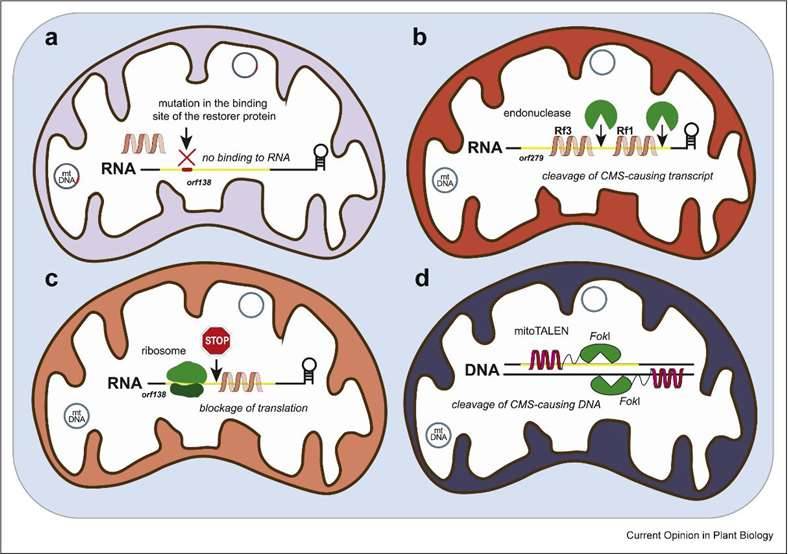 Mitochondrial-nuclear genome interactions in CMS and fertility restoration in plants. (Melonek et al., 2022)
Mitochondrial-nuclear genome interactions in CMS and fertility restoration in plants. (Melonek et al., 2022)
Comparative investigations into reference genome sequences and pan-genomic data have unveiled a substantial proliferation and extraordinary diversity within the RFL and mTERF gene families, which serve as the primary reservoirs of fertility restoration genes in cereal crops. Looking ahead, the integration of genome-wide association studies with advanced long-read sequencing technologies is poised to expedite the identification and characterization of novel restoration genes, poised to enhance hybrid breeding programs. Furthermore, these breakthroughs promise to yield fresh insights into the mechanisms by which nuclear Rf genes effectively mitigate CMS-related traits.
Reference:
- Melonek, Joanna, and Ian Small. "Triticeae genome sequences reveal huge expansions of gene families implicated in fertility restoration." Current Opinion in Plant Biology 66 (2022): 102166.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


