CD Genomics provides statistical and bioinformatic data analysis services that help our customers to explain large amounts of data generated by sequencing, genotyping, and microarray experiments.
Overview
Next-generation sequencing has fundamentally revolutionized biological and medical sciences. A sequencing/microarray data analysis project requires careful planning, seasoned methodology, and concise reporting. CD genomics is an expert company offering comprehensive bioinformatics solutions in all aspects of sequencing and microarray. We provide data analysis services for data generated by a wide range of sequencing/microarray platforms. We use the cutting-edge bioinformatics algorithms and pipeline, and provide you with high-quality and ready-for-publication figures.
Our bioinformatic staff consists of PhD-level scientists trained in computer languages, statistics, bioinformatics, genetics, and genomics. The software infrastructure for analysis is a combination of custom-built and open-source software. Our bioinformatics services take your raw next/third generation data and provide you with comprehensive figures customized to your research purposes as well as personalized data interpretation support. CD Genomics’ full range of bioinformatics services can be an ideal solution for you.
Bioinformatics Workflow
High-throughput sequencing technology can generate numerous data with heterogeneous quality, and there is no optimal bioinformatics pipeline for all cases. We are specialized in customizing the bioinformatics pipeline for your specific projects. We support companies and research groups by turning ideas into concrete project plans and implementing them, thereby making full use of your data.
- NGS data analysis workflow
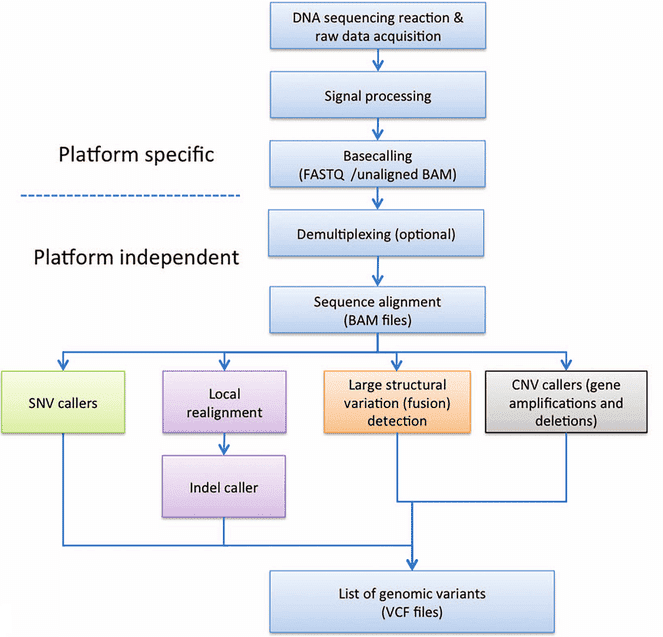 (Roy et al. 2016)
(Roy et al. 2016)
|
General bioinformatics workflow for NGS data includes base calling (platform specific), demultiplexing (optional), sequence alignment, and variants calling. |
- Microarray data analysis workflow
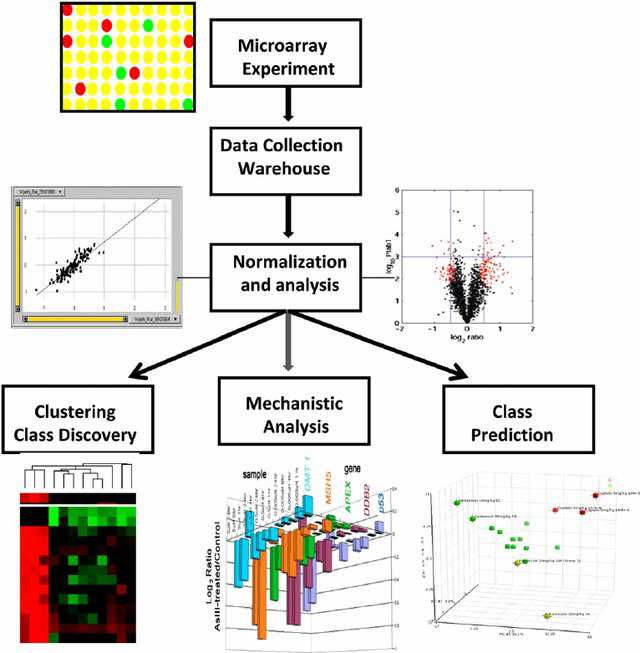 (Afshari et al. 2010)
(Afshari et al. 2010)
|
General bioinformatics workflow for microarray data includes data correction for background, normalization, gene expression patterns clustering, class prediction, and biological mechanism prediction. |
Service Range
CD Genomics' bioinformatics services cover a wide range of genomics applications, including but not limited to genomics, transcriptomics, epigenomics, and microarray.
| Genomic Data Analysis (targeted, exome and whole genome sequencing) |
Metagenomics |
Transcriptomic Data Analysis |
Epigenomics Data Analysis |
Long-Read Sequencing Data Analysis |
Single-cell RNA Sequencing Data Analysis |
Microarray Data Analysis |
| 1. Disegno in silico of gene panel |
1. RNA16S sequencing |
1. Sequence quality checks |
1. Analysis of ChIP-Seq data (transcription factors, histones) |
1. Quality Control and Error Correction |
1. Quality Control and Preprocessing |
1. Microarray data quality control |
| 2. Sequence quality checks |
2. Identification of microbial communities composition |
2. De-novo transcriptome assembly |
2. Analysis of bisulfite sequencing data (Methyl-Seq) |
2. Gene/Gene Isoform Expression |
2. Exploratory Analysis |
2. Differential expression analysis |
| 3. De-novo genome assembly |
|
3. Differential expression analysis of genes, isoforms and exons |
3. Differential methylation analysis in CpG and non-CpG regions |
3. Novel Gene Discovery and Full-Length Isoform Identification |
3. Cell Type Identification |
|
| 4. Sequence alignment versus reference genomes |
|
4. Analysis of fusion genes, circularized RNAs and trans-splicing events |
4. HiC analysis |
4. De Novo Fusion Gene Detection and Fusion Isoform Expression Profiles |
4.Trajectory Analysis |
|
| 5. Variant calling, annotation and prioritization (single samples, matched normal-tumor pairs, family trios) |
|
5. Detection of long non-coding RNAs |
|
5. Allele-Specific Expression and Haplotyping |
5.Integrative Single-cell Analyses |
|
| 6. Gene-ontology and pathway enrichment analysis |
|
6. Gene-ontology and pathway enrichment analysis |
|
6. De Novo Genome Assembly |
6. Ligand-Receptor Analysis |
|
| 7. Analysis of CNV and large rearrangements |
|
7. Gene expression-dependent survival analysis |
|
7. De Novo Transcriptome Assembly |
7. Spatial Transcriptomic Analysis |
|
| 8. Clonal evolution and heterogeneity in cancer |
|
|
|
8. Methylation Calling, Nucleosome Positioning, and Chromatin Accessibility |
We Have Two Options For You:
Standard analysis: Results are published without the service providing any additional context for the experiment (e.g., list of variations with annotations, genes differentially expressed with statistical data, binding sites, etc.). In this situation, it is anticipated that the sequencing cost will be increased by a typical cost per sample.
advanced analysis. Here, the objective is to examine the facts while also giving them context through expert guidance. In fact, the service works with the study's design, creates ad hoc analyses for the study, recommends the best way to present the data, etc.
Our advantages
- Custom bioinformatics
- Illumina, Ion Torrent, PacBio SMRT, and Nanopore compatible
- Publication-ready data and figures
- Accurate sequencing and microarray services available
- A professional and experienced expert team
- Fast turnaround time
Talk to a scientist
Want more information about our bioinformatics services or interested in bioinformatics analysis of your raw high-throughput sequencing or microarray data? Please feel free to submit a service inquiry. We are here to help!
References:
- Roy, S.; et al. Next-Generation Sequencing Informatics: Challenges and Strategies for Implementation in a Clinical Environment. Archives of Pathology & Laboratory Medicine, 2016, 140(9), 958–975.
- Afshari, C. A., Hamadeh, H. K., & Bushel, P. R. The Evolution of Bioinformatics in Toxicology: Advancing Toxicogenomics. Toxicological Sciences, 2010, 120(Supplement 1), S225–S237. doi:10.1093/toxsci/kfq373
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


