In 2021, on the 20th anniversary of the release of the human genome sequence sketch, the Telomere-to-Telomere (T2T) consortium announced the latest complete human genome sequence, CHM13v1.1, which not only contains all unresolved sequences but also corrects the original assembly errors, making it the most complete human genome sequence to date. The results include the seamless assembly of all 22 human autosomes and X chromosomes except for the Y chromosome, completing a challenging task that 8% of the Human Genome Project has yet to solve.
Background
Telomeres are the terminal part of eukaryotic linear chromosomes, a special structural region that plays an important role in the structure and stability of linear chromosomes. Telomere DNA is composed of simple highly repetitive sequences of DNA that are difficult to assemble.
The most recent version of the human genome atlas was released in 2013 before the release of the complete human genome and was called GRCh38. Since then, it has been repeatedly patched. To date it still has gaps, missing more than 8% of the genome, including all of the satellite arrays of the mitotic regions, telomeres, large repeats of the genome, and rRNA regions that have long been in an unknown or unknowable sequence state. These missing genomes are hidden in long sequences with a large number of repetitive gene copies, which cannot be perfected by short read length and assembly techniques.
With Pacific Biosciences (PacBio) HiFi and Oxford Nanopore Technologies (ONT), long-read sequencing technologies overcoming the low throughput of Sanger sequencing and the short read length of short-read sequencing technologies, combined with the continuous optimization of assembly algorithms, more and more perfect genomes of model organisms have been published. genomes are being published.
Complete T2T Genome Map
Using ONT ultra-long and PacBio HiFi sequencing data with high precision and long sequences, researchers from the T2T consortium have assembled and published the first human X chromosome completion map, autosome completion map, and human genome completion map, and the related results have been published in Science and other journals.
The purpose of T2T genome is to obtain a high-quality genome with high accuracy, continuity and completeness from telomere to telomere. The researchers completed the assembly of the genome at the chromosome level by combining Hi-C technology to obtain information on the relative positions of genes on the chromosomes, and manual adjustment for complex regions to obtain the T2T reference genome sequence.
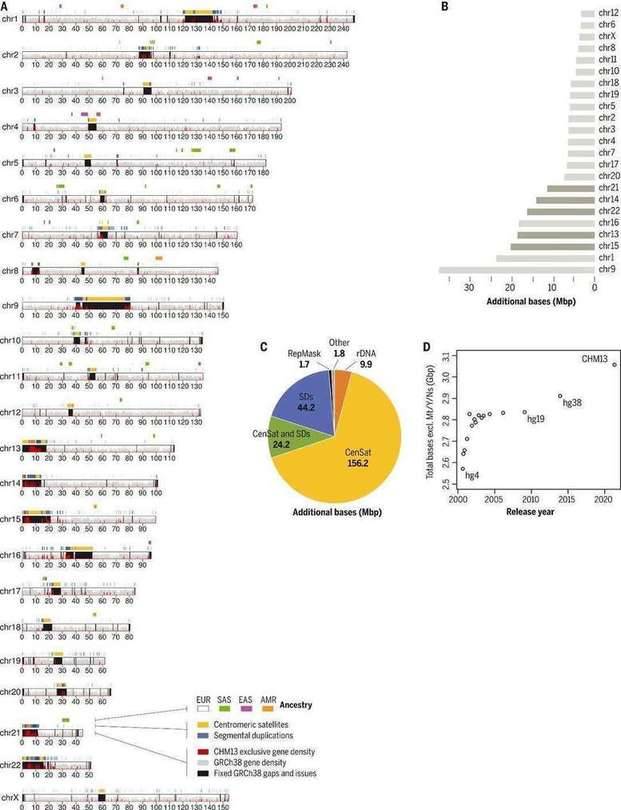 The complete T2T-CHM13 human genome assembly (Nurk S et al., 2022)
The complete T2T-CHM13 human genome assembly (Nurk S et al., 2022)
In this completed human genome map, the investigators added or corrected 238 Mb of new sequences, of which 182 Mb were completely new, and annotated to 2,226 new genes. As a result, the results eliminate tens of thousands of false positive variants in each sample, including reducing false positives for 269 medically relevant gene tests by more than 90%.
In addition to the human telomere-to-telomere genome, the complete T2T genome sequence of Arabidopsis has been assembled. Using PacBio HiFi and ONT ultra-long data, a near-completion map of Arabidopsis containing five tethers was stitched together: chromosomes 1, 3 and 5 contain the complete telomere-to-telomere sequence, while 2 and 4 remain unassembled in the 45s rDNA region of the short arm and the adjacent telomeric region. Through identification and sequence analysis, the researchers reveal a model for the recombination-based homogenization process in Arabidopsis, advancing the study of the structure and function of the tethers, as well as the evolutionary mechanisms.
Significance
T2T genome assembly is very difficult, and it is still difficult to read through regions with long repetitive sequences as well as mitotic regions in some species by current sequencing and data analysis technologies. The present human genome assembly circumvents some complexity by using a haploid cell line with human staph extensions in order to avoid sequencing 2 different X chromosomes. Since then, the construction of a more perfect reference genome containing T2T, haplotype information and sex chromosome information will become a research trend in genomics. And high quality T2T reference genome sequences can help to reveal more important genetic information about diseases, aging, evolution and other important life processes.
Recommended Services
CD Genomics offers Human Whole Genome PacBio SMRT Sequencing and Whole Genome Sequencing based on Illumina, Nanopore sequencing and PacBio SMRT sequencing platforms. We provide comprehensive genome sequencing services including sequencing, assembly, and data analysis for a variety of species.
References:
- Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science, 2022, 376(6588): 44-53.
- Logsdon, G. A. et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021).
- Naish, M. et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374, eabi7489 (2021).
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


