Human Mitochondrial Genome
Mitochondria, essential organelles found in all eukaryotic cells, are central to energy production through aerobic respiration and play key roles in cellular processes. They possess their own unique genetic material, the mitochondrial genome (mtDNA).
Human mtDNA is a circular molecule, spanning 16,569 base pairs, consisting of heavy and light strands. It is predominantly a coding region, encompassing 37 genes, including 22 tRNAs, 2 rRNAs, and 13 proteins crucial for mitochondrial electron transport chain components. Unlike nuclear DNA, mtDNA follows a strict matrilineal inheritance pattern.
Within cells, mitochondria are numerous, each containing multiple mtDNA copies. This multicopy nature leads to genetic diversity, with mutation percentages ranging from 0% to 100%. Changes in mutation load during cell division can impact energy production, causing a "threshold effect" and cellular dysfunction.
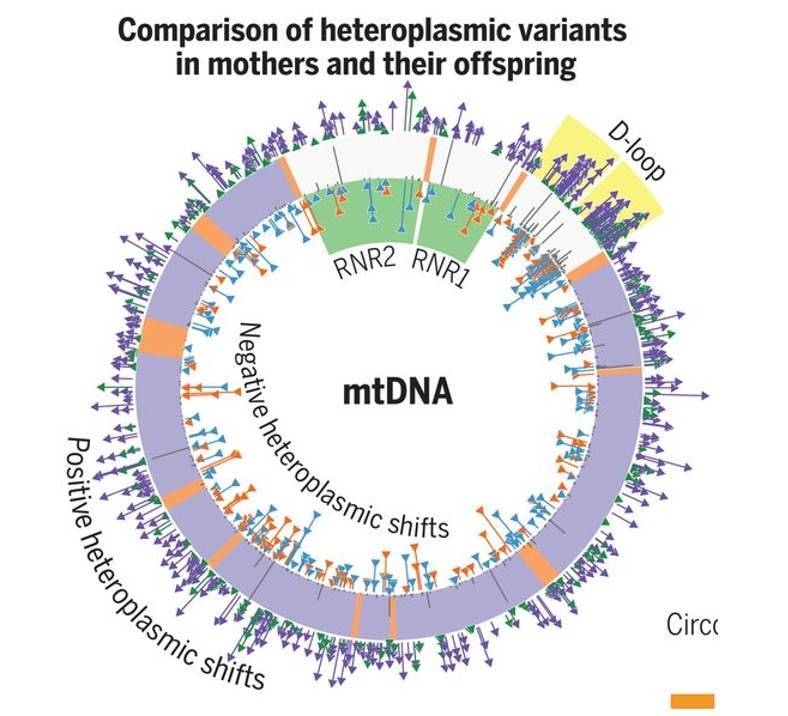 Germline selection shapes human mitochondrial DNA diversity. (Wei et al., 2019)
Germline selection shapes human mitochondrial DNA diversity. (Wei et al., 2019)
High-throughput Sequencing of Mitochondrial Genomes
In both clinical and scientific studies, there is a growing preference for employing advanced techniques like Next-Generation Sequencing (NGS), encompassing targeted panel sequencing, whole mitochondrial genome sequencing, Whole Exome Sequencing (WES), and Whole Genome Sequencing (WGS), for the comprehensive analysis of mitochondrial genomes. By adopting these methodologies, not only is the analytical efficiency significantly heightened, but the sensitivity of pathogenic variant detection is also markedly enhanced, primarily due to the augmentation of sample sizes.
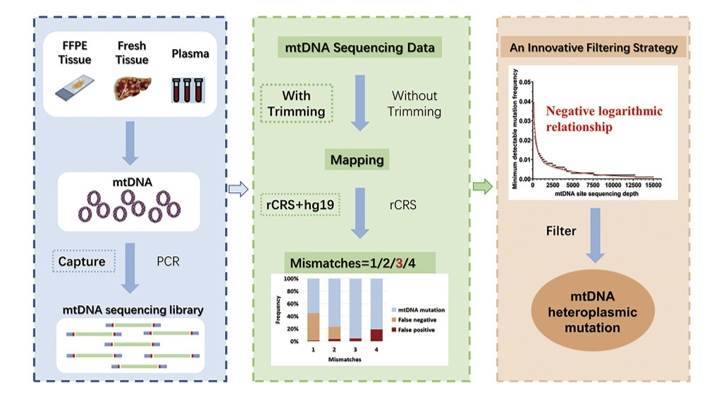 Accurate next-generation sequencing detection of tumor mitochondrial DNA mutations. (Guo et al., 2020)
Accurate next-generation sequencing detection of tumor mitochondrial DNA mutations. (Guo et al., 2020)
- Targeted Panel Sequencing
In the initial stages of Next-Generation Sequencing (NGS) for mitochondrial diseases, only specific segments of the mitochondrial genome are sequenced. These segments encompass peptides encoding the respiratory chain and well-known genes associated with disease. Alternatively, we can select all mitochondrial-related genes, including both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), as listed in MitoCarta. This comprehensive panel is commonly referred to as the "MitoExome."
- Whole Mitochondrial Genome Sequencing
Utilizing NGS for whole mitochondrial genome sequencing allows us to detect any mitochondrial DNA (mtDNA) variants and provides a precise assessment of variant heterogeneity. Prior to embarking on Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS), many mitochondrial diagnostic centers often perform mtDNA sequencing. This serves the dual purpose of identifying potentially pathogenic mtDNA variants and ruling out their presence. It's worth noting that many pathogenic mtDNA variants are exclusively found in clinically affected tissues, such as skeletal muscle.
Essentially, Whole Exome Sequencing (WES) involves the targeted sequencing of exonic regions within the genome. However, a substantial portion of WES data is generated from non-targeted regions, including the mitochondrial genome. Given that the mitochondrial genome consists of thousands of copies, in contrast to the diploid nuclear genome, there are significantly more off-target mtDNA sequences than off-target nDNA sequences (by a factor of 30-120 times). Notably, the proportion of captured mtDNA sequences is correlated with the abundance of mtDNA in the original total DNA sample. For instance, individual cells in the heart and skeletal muscle contain a higher mtDNA content than peripheral blood. The high copy number nature of mtDNA results in a mixture of wild-type and mutant mtDNA in proportions, known as heterogeneity, which can theoretically range continuously from 0% to 100%. Furthermore, pathogenic mtDNA mutations are typically highly heterozygous or homozygous in carriers, but they exhibit low heterozygosity in cases of asymptomatic pathogenic mtDNA mutations. Conversely, mtDNA copy number is known to be highly variable and is associated with various diseases, including cancer. Hence, mtDNA copy number is a crucial metric for mitochondrial genome analysis. As the coverage of sequencing data is directly proportional to the number of chromosomes, it is possible to estimate mtDNA copy number from WES data.
WGS offers the capability to detect all genetic variants across the entire human genome, significantly enhancing the diagnostic potential for mitochondrial diseases. Some studies have shown a high degree of concordance between traditional PCR-based methods and WGS data for analyzing mtDNA variants. Additionally, when it comes to assessing copy number, mtDNA copy number estimates derived from WGS data tend to be more reliable.
Bioinformatic Analysis of Mitochondrial Genomes
- Detection of Mitochondrial Variants
Presently, the consistency in software and processes used to identify mitochondrial variants across various research articles is lacking. Several software tools have been developed for the analysis and detection of mitochondrial variants from high-throughput sequencing data, such as mtDNA-Server, MitoSeek, MToolBox, among others. For comprehensive guidance on analyzing mitochondrial genomes, it's worthwhile to refer to the recommended procedures provided by GATK, as detailed here.
Moreover, when analyzing mitochondrial genes using Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) data, the variant quality control process closely resembles that of the nuclear genome. It is essential to pay attention to referencing the genome version when calculating sequencing depth. Note that in the human hg19 reference genome, the mitochondrial genome is represented as CRS (NC_001807), but the current mainstream human mitochondrial reference genome is rCRS (NC_012920).
- Assignment of Mitochondrial Haplogroups
Presently, the most commonly utilized software for assigning mitochondrial haplogroups to samples is HaploGrep2. HaploGrep2 assigns the mitochondrial haplogroups to each sample based on PhyloTree17, with rCRS serving as the reference genome. This software also enables direct input of a variant call file (vcf file) to determine the most suitable mitochondrial haplogroup for each sample, based on the mitochondrial variants detected within each sample. Another viable option for haplogroup assignment is MitoTool.
- Estimation of Mitochondrial Copy Number
During Next-Generation Sequencing (NGS), both autosomal and mitochondrial chromosomes have an equal likelihood of being detected. Consequently, the average coverage of autosomal and mitochondrial chromosomes is theoretically proportional to their copy number.
Mitochondrial Variant Databases
In contrast to nuclear DNA (nDNA), the availability of population databases for mitochondrial DNA (mtDNA) variants is considerably limited. Fortunately, the practice of reanalyzing mtDNA sequences from existing Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) datasets is widely embraced and employed. Several substantial population-level mitochondrial databases have emerged, including Mitomap, HmtDB, 1000 Genomes, gnomAD, and MitImpact.
Mitomap stands as the preeminent choice for mtDNA analysis. Beyond serving as a repository of mtDNA variants across populations, it has evolved into a pivotal resource for the mitochondrial research community. With decades of recognition, Mitomap updates its dataset by extracting mtDNA sequences from GenBank biannually and subjecting them to rigorous bioinformatic analysis. As of July 1, 2022, Mitomap encompasses 56,910 complete mtDNA sequences and 78,504 mtDNA control region sequences, including 19,449 mtDNA single nucleotide polymorphisms (SNPs).
The gnomAD (Genome Aggregation Database), the largest open-source human variation database, made mtDNA variants publicly accessible for the first time in November 2020 with gnomAD v3.1, comprising a total of 56,434 mtDNA sequences from WGS data, housing 10,850 mtDNA variants.
Conversely, the HelixMTdb database is founded on WES data from 196,554 unrelated individuals, detecting a total of 15,035 mitochondrial variants. Notably, a predominant portion of these samples originate from European populations.
HmtDB is another noteworthy database, encompassing samples with both healthy and disease-related phenotypes. It is meticulously designed to support population genetics and assist clinicians in assessing the pathogenicity of specific mtDNA variants. It's worth highlighting that both HmtDB and Mitomap source their mtDNA sequences from GenBank, leading to significant overlap in sample sources between these two databases.
Additionally, some of the widely-used disease databases for nuclear DNA variants, including ClinVar, Clinvar Miner, and OMIM, not to mention Mitomap and HmtDB, also provide information regarding the clinical implications of mtDNA variants. As of July 1, 2022, Mitomap has compiled data on 455 rRNA/tRNA mutations and 545 coding/non-coding variants associated with diseases, of which only 97 variants have been definitively categorized as pathogenic.
Tools for Predicting the Impact of Mitochondrial Variants
Numerous tools designed for assessing the deleteriousness of variants within the nuclear DNA (nDNA) coding regions can also be applied to variants within the mitochondrial DNA (mtDNA) coding regions. Prominent among these tools are CADD, PolyPhen-2, SIFT, Mutpred, and PROVEAN, several of which are extensively utilized for prioritizing nDNA variants. However, it is important to note that some of these tools, originally tailored for nDNA coding regions, may exhibit reduced accuracy when applied to mtDNA coding regions.
| Tool |
Applicability |
Key Features |
| Common Tools for Assessing mtDNA and nDNA Variants |
Both mtDNA and nDNA coding region variants |
- CADD, PolyPhen-2, SIFT, Mutpred, and PROVEAN. |
| - Originally designed for nDNA, may have reduced accuracy for mtDNA. |
| Specialized mtDNA Tools with Better Performance |
Specifically designed for mtDNA |
- MToolBox |
| - APOGEE |
| - Mitoclass |
| Tools for Mitochondrial tRNA Variants |
Mitochondrial tRNA variants |
- MitoTIP |
| - PON-mt-RNA |
| MToolBox |
Mitochondrial variant analysis |
- Analyzes human mitochondrial sequences from NGS. |
| - Provides mitochondrial variant heterogeneity annotation. |
| - Assists in the identification of pathogenic variants. |
References:
-
Wei, Wei, et al. "Germline selection shapes human mitochondrial DNA diversity." Science 364.6442 (2019): eaau6520.
- Guo, Shanshan, et al. "An innovative data analysis strategy for accurate next-generation sequencing detection of tumor mitochondrial DNA mutations." Molecular Therapy-Nucleic Acids 23 (2021): 232-243.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


