Overview of T-cell Receptors (TCRs)
Human adaptive immune system drives the immune response via B cell-generated immune-globulins and immunoglobulin-like TCRs on T lymphocytes. TCRs are responsible for the recognition of the Ag-MHC (major histocompatibility complex) molecules. TCRs normally consist of highly variable α and β chains expressed by a majority of T cells, or γ and δ chains expressed by a subset of T cells (1–5%). The TCR chains consist of a variable region for antigen recognition and a constant region. The variable region of α and δ chains is encoded by variable (V) and joining (J) genes, while β and γ chains are encoded by diversity (D) genes. Each TCR chain contains three hypervariable loops, i.e., complementarity determining regions (CDR1–3). The antigen specificity of TCRs is mainly determined by the hypervariable CDR3 of the beta chain, which is formed by recombination of V, D, and J gene segments with the addition of random nucleotides at the gene segment junctions. The sum of all TCRs of a human body is termed the TCR repertoire or TCR profile, which can change greatly with the onset and progression of diseases. Hence, it is more important to determine the immune repertoire status under different disease conditions.
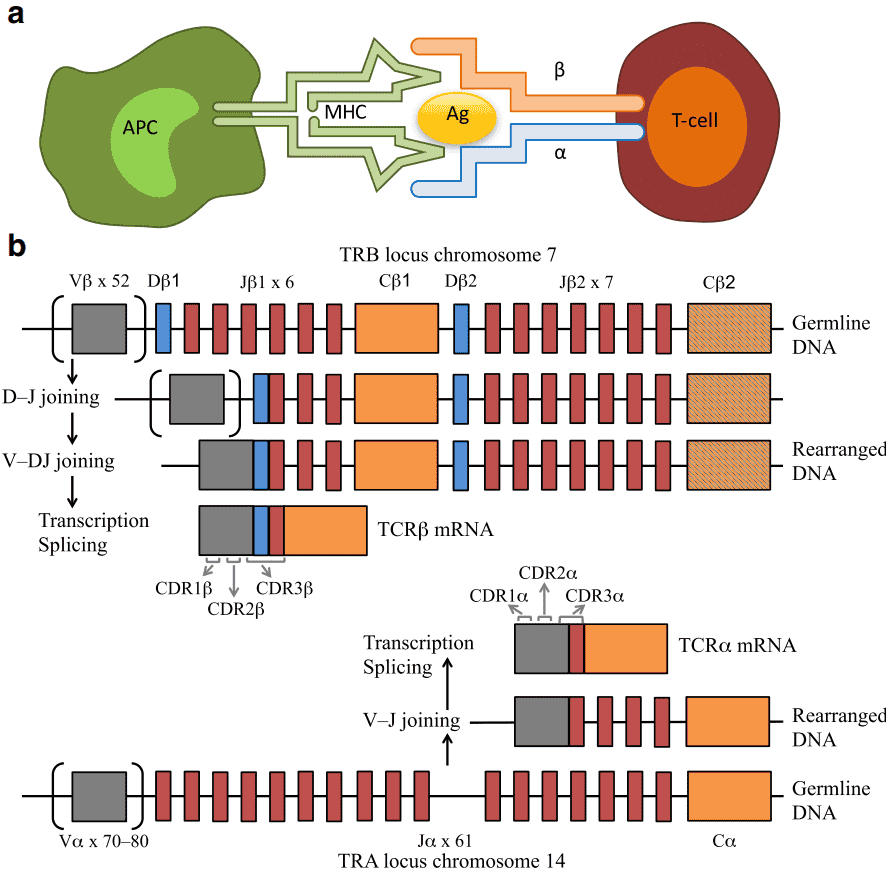 Figure 1. Interaction between an antigen-presenting cell (APC) and a T cell (a), and V(D)J recombination (b) (Rosati et al., 2017).
Figure 1. Interaction between an antigen-presenting cell (APC) and a T cell (a), and V(D)J recombination (b) (Rosati et al., 2017).
TCR Sequencing
Next-generation sequencing (NGS) has revolutionized TCR repertoire profiling through the ability to massively analyze TCR and antibody repertoires from blood samples. We mainly focus on cell population-based TCR sequencing in this review.
γδ T cells are less interesting since they are frequently found at mucosal sites. The β chain is the main target of interest because of its uniqueness in single cells and higher combinatorial potential compared to α chains. PCR-based methods can amplify both α and β chains simultaneously, but they are often separated during sequencing in order to increase the precision and specificity of the outcome.
The CDR3 region has been a preferential target in many TCR profiling studies. Although CDR1 and CDR2 do not directly interact with antibodies, they play a vital role contacting the MHC molecules and thus affecting the sensitivity and affinity of the TCR binding.
There are three common methods for the construction of TCR profiling libraries: multiplex PCR, target enrichment, and 5'RACE cDNA synthesis and nested PCR (Figure 2). Molecular barcodes have been introduced during cDNA synthesis to minimize the impact of PCR and sequencing errors.
Among them, multiplex PCR approaches are the most common and can be used for both gDNA and RNA. Generally, primers for the J alleles or the constant region of the α and β chains, as well as primers for all known V alleles are used to amplify TCR transcript across the CDR3 region. However, this method cannot detect novel V alleles variants and can introduce amplification biases.
Both Agilent (SureSelectXT) and Illumina (TruSeq) offer targeted enrichment-based kits for capturing TCRs of αβ T cells. RNA or gDNA are pre-processed and then incubated with custom-designed RNA baits to hybridize with the gDNA/cDNA target, followed by a further amplification step. Target enrichment methods are less susceptible to PCR bias.
For RNA samples, rapid amplification of 5' complementary DNA ends (5'RACE) based on template switch is becoming a gold standard for bulk TCR studies. Marketed by Clonotech as "SMART" technology, this method enables enrichment of all TCR variants in the sample. Clonotech has developed a commercial kit for TCR studies. cDNA synthesis is performed using primers against the constant region of the TCR mRNA transcript. Subsequently, a nested PCR is carried out to amplify the constant region.
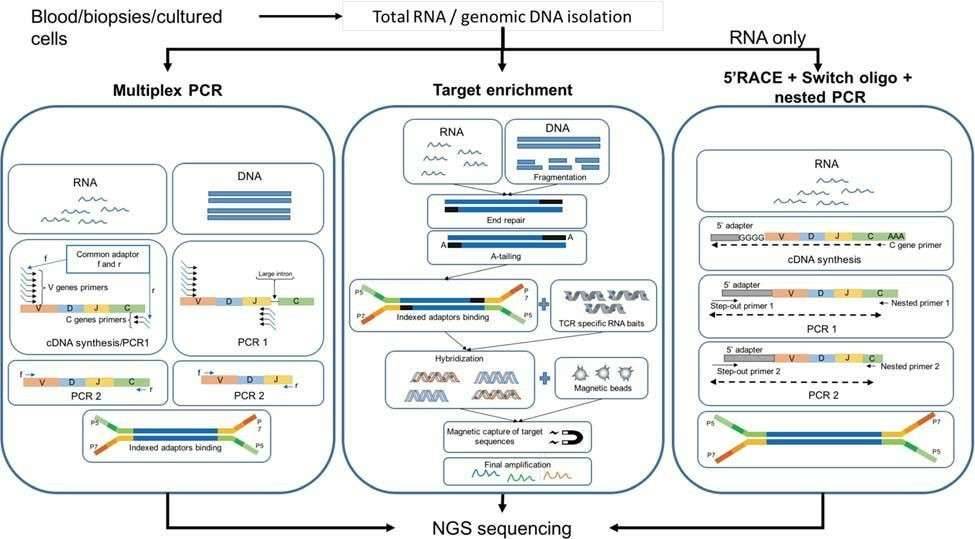 Figure 2. Workflow of three principal methodologies for TCR library preparation (Rosati et al., 2017).
Figure 2. Workflow of three principal methodologies for TCR library preparation (Rosati et al., 2017).
Deep sequencing allows the acquisition of a more complete TCR repertoire. However, for disease-oriented studies that look for highly expressed TCRs, a superficial low-coverage sequencing may be sufficient. In this case, Illumina MiSeq is commonly used, while Illumina HiSeq is more commonly used for deep sequencing.
TCR Profiling from RNA-Seq Data
As transcriptome sequencing becoming routinely used in basic and clinical studies, it could serve as an important source for TCR profiling. Several groups have developed tools for TCR repertoire extraction from bulk or single-cell RNA-seq data, such as MiXCR. This tool aims to extract as many CDR3 sequences as possible, hardly without false positives.
References:
- Bolotin D A, Mamedov I Z, Britanova O V, et al. Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms. European journal of immunology, 2012, 42(11): 3073-3083.
- Bolotin D A, Poslavsky S, Davydov A N, et al. Antigen receptor repertoire profiling from RNA-seq data. Nature biotechnology, 2017, 35(10): 908.
- Hsu M S, Sedighim S, Wang T, et al. TCR sequencing can identify and track glioma-infiltrating T cells after DC vaccination. Cancer immunology research, 2016, 4(5): 412-418.
- Rosati E, Dowds C M, Liaskou E, et al. Overview of methodologies for T-cell receptor repertoire analysis. BMC biotechnology, 2017, 17(1): 61.
- Luo W, Cui J H, Lin K R, et al. TCR repertoire as a novel indicator for immune monitoring and prognosis assessment of patients with cervical cancer. Frontiers in immunology, 2018, 9: 2729.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


