
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

CRISPR/Cas9 technology is one of the most popular methods used for genome editing by introducing both Cas9 endonuclease and a guide RNA into the cells of interest. The guide RNA is designed to direct the Cas9 endonuclease to a particular site in the genome where it produces a double-strand break (DSB). There are generally two ways to repair the double-stranded break: non-homologous end-joining (NHEJ) or homologous directed repair (HDR). NHEJ is the main form of repair in mammalian cells. As it is error-prone, repair via the NHEJ pathway allows for insertion, deletion, loss-of-function mutations which probably result in frameshifts and affect protein expression. In addition to knockout mutations, a template DNA can be introduced to direct HDR and create mutations in the gene-of-interest. HDR faithfully copies the template sequence to the cut target site.
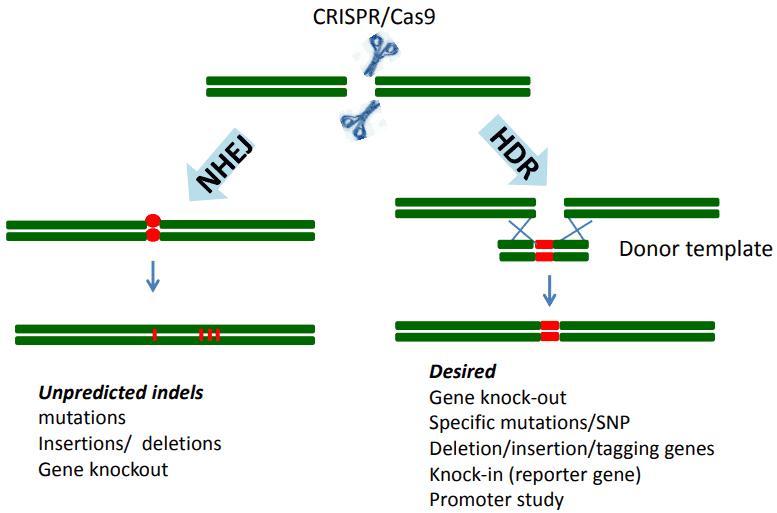 Figure 1. Genome editing by CRISPR/Cas9 technology is achieved via repair.
Figure 1. Genome editing by CRISPR/Cas9 technology is achieved via repair.
You may interested in
Although the CRISPR system is efficient for genome editing. However, some cells in a population will not be edited, some will have one allele edited, and some will have both alleles edited. It is important to validate genome edits after CRISPR/Cas9 experiments. Next generation sequencing (NGS), as a powerful and high-throughput approach, can be utilized for screening of CRISPR-induced mutations. NGS can simultaneously look at off-target changes in a large number of samples. When using this method, it is necessary to keep a set of control cells. Software such as CRISPResso can be used for data analysis. NGS is also suited for assessing genome edits created by ZFN (Zinc-finger nuclease) or TALEN (Transcription activator-like effector nuclease).
Sentmanat et al. (2018) described the NGS approach for genome edits validation in his published work. Briefly, CRISPR sequencing involves a two-step PCR protocol and deep sequencing. First, the target genomic site of interest is amplified with a primer that contains partial Illumina sequencing adaptors. Next, a second PCR with primers containing indices and necessary Illumina sequencing adaptors. As a result, the target regions will be amplified. The qualified PCR products are then subjected to deep sequencing with the Illumina MiSeq platform.
NGS techniques for validation encompass several key approaches:
Whole-Genome Sequencing (WGS) stands out as a comprehensive method for discerning CRISPR/Cas9-induced genomic changes. By sequencing the entire genome, WGS offers unparalleled coverage to identify insertions, deletions, and other alterations. This technique is instrumental in assessing the effectiveness of CRISPR/Cas9 editing, while also uncovering rare mutations within the genome.
Targeted Amplicon Sequencing, on the other hand, zooms in on specific genomic regions, including CRISPR/Cas9 target sites and neighboring areas. With its high depth of coverage, this technique excels at detecting minutiae mutations, serving as a valuable tool to corroborate the precision of CRISPR/Cas9 alterations. Particularly beneficial for assessing edits in individual genes or a limited set of targets.
Off-Target Analysis is pivotal in predicting and sequencing potential off-target sites generated by CRISPR/Cas9 modifications. This method aids in evaluating the specificity and safety of CRISPR/Cas9 edits by identifying unintended alterations. It plays a crucial role in assessing the accuracy and safety profiles of CRISPR/Cas9 editing methodologies.
Long-Range PCR and Sequencing take a complementary approach by amplifying large genomic regions surrounding CRISPR/Cas9 target sites before sequencing. This method is adept at capturing structural variations and chromosomal rearrangements stemming from CRISPR/Cas9 manipulations. It proves to be a valuable tool in detecting substantial genomic changes induced by CRISPR/Cas9 editing procedures.
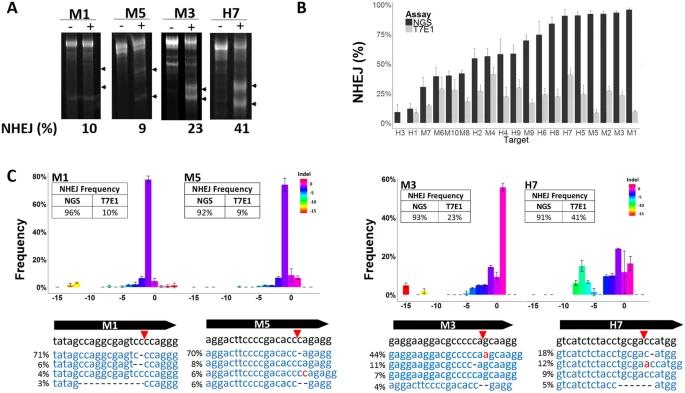 Figure 2. CRISRP-Cas9 activity reported with the T7E1 Assay and Next-Generation Sequencing. (Sentmanat et al., 2018)
Figure 2. CRISRP-Cas9 activity reported with the T7E1 Assay and Next-Generation Sequencing. (Sentmanat et al., 2018)
Another limitation of CRISPR technology is the occurrence of Off-target cuts. The CRISPR system cuts not just at its target place, but also at unintended sites with similar sequences. These off-target cuts may produce undesirable and even harmful mutations. Over the past few years, scientists have established several NGS-based approaches to detect off-target mutations.
Table 1. NGS-based approaches to detect off-target mutations.
| Assays | Description | Resources |
| In vitro genome-wide assays | ||
| Digenome-Seq | Genomic DNA is first digested with a nuclease and then subjected to whole genome sequencing. Off-targets can be computationally identified. | Kim et al. 2015 Web tool: http://www.rgenome.net/digenome-js/#! Code: https://github.com/chizksh/digenome-toolkit2 |
| SITE-Seq | Genomic DNA is cleaved using Cas9 nuclease, and Cas9 nuclease cleavage sites are biochemically tagged and enriched. High-throughput sequencing and bioinformatics analysis is then used to detect off-target cleavage sites. | Cameron et al. 2017 Protocol: Protocol Exchange, doi:10.1038/protex.2017.043 |
| Cell-based genome-wide assays | ||
| LAM-HTGTS | Chromosomal translocations of off-target and on-target breaks are PCR amplified and analyzed by NGS. | Frock et al. 2015 Protocol: Nat Protoc, 11:853-71, 2016 Code:http://robinmeyers.github.io/transloc_pipeline/index.html |
| BLISS | DSBs are biochemically labeled, and their downstream sequences are PCR amplified and analyzed using NGS. | Yan et al. 2017 |
References:


