We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
circRNA Full-Length Identification and Quantification Service
Circular RNAs (circRNAs) have emerged as significant regulators in gene expression with potential therapeutic applications. Since their discovery through high-throughput sequencing in 2012, the understanding and exploration of circRNAs have dramatically increased. Currently, studies involving circRNA are predominantly conducted utilizing next-generation sequencing (NGS) platforms. Although NGS can simultaneously sequence millions of short-read sequences, it is inadequate for capturing the full length of circRNA and presents challenges in the accurate quantification of these molecules.
Researchers focusing on circRNA are well aware of the inherent limitations associated with NGS in detecting circRNA. The primary issues include the inability to capture the complete length of circRNA and the difficulty in precise quantification. Although both experimental and bioinformatic strategies have been developed to mitigate these deficiencies, the inherent short-read length of NGS remains a fundamental obstacle that cannot be entirely overcome.
CD Genomics offers an advanced circRNA full-length identification and quantification service utilizing third-generation sequencing (TGS) technologies, addressing these limitations and providing comprehensive insights into circRNA biology.
Challenges and Limitations of circRNA Short-Read Sequencing
Experimental Aspects:
CircRNA enrichment can be achieved to some extent using RNase R or poly(A)-based library construction methods. However, certain circRNAs exhibit sensitivity to RNase R, while some linear molecules can resist RNase R digestion. Additionally, PCR amplification and library complexity may introduce biases in circRNA quantification.
Contemporary research indicates that the majority of circRNAs have lengths predominantly distributed between 300 to 500 base pairs (bp). Consequently, modifying sequencing strategies from paired-end 150 bp reads to paired-end 250 bp reads can enhance the sensitivity and accuracy of circRNA detection.
Bioinformatics Aspects:
Current methods for circRNA identification and quantification predominantly rely on back-spliced junctions (BSJs). There are also alternative approaches, including the following:
Combining forward-spliced junctions (alignment to linear splice sites) for the relative quantification of circRNAs.
Similar to linear RNA sequence reconstruction, circRNA identification involves stitching reads together and utilizing deep search algorithms to identify the optimal transcript strategy.
These methodologies improve the detection and quantification of circRNAs, yet the fundamental limitation of short-read sequences remains a significant challenge.
circRNA Full-Length Sequencing Service
As research on circRNA progresses, the accurate identification and comprehensive analysis of circRNA full-length sequences have become increasingly critical. However, the effective analysis and acquisition of these sequences remain complex and warrant further investigation. The question arises whether an innovative strategy can directly achieve this objective.
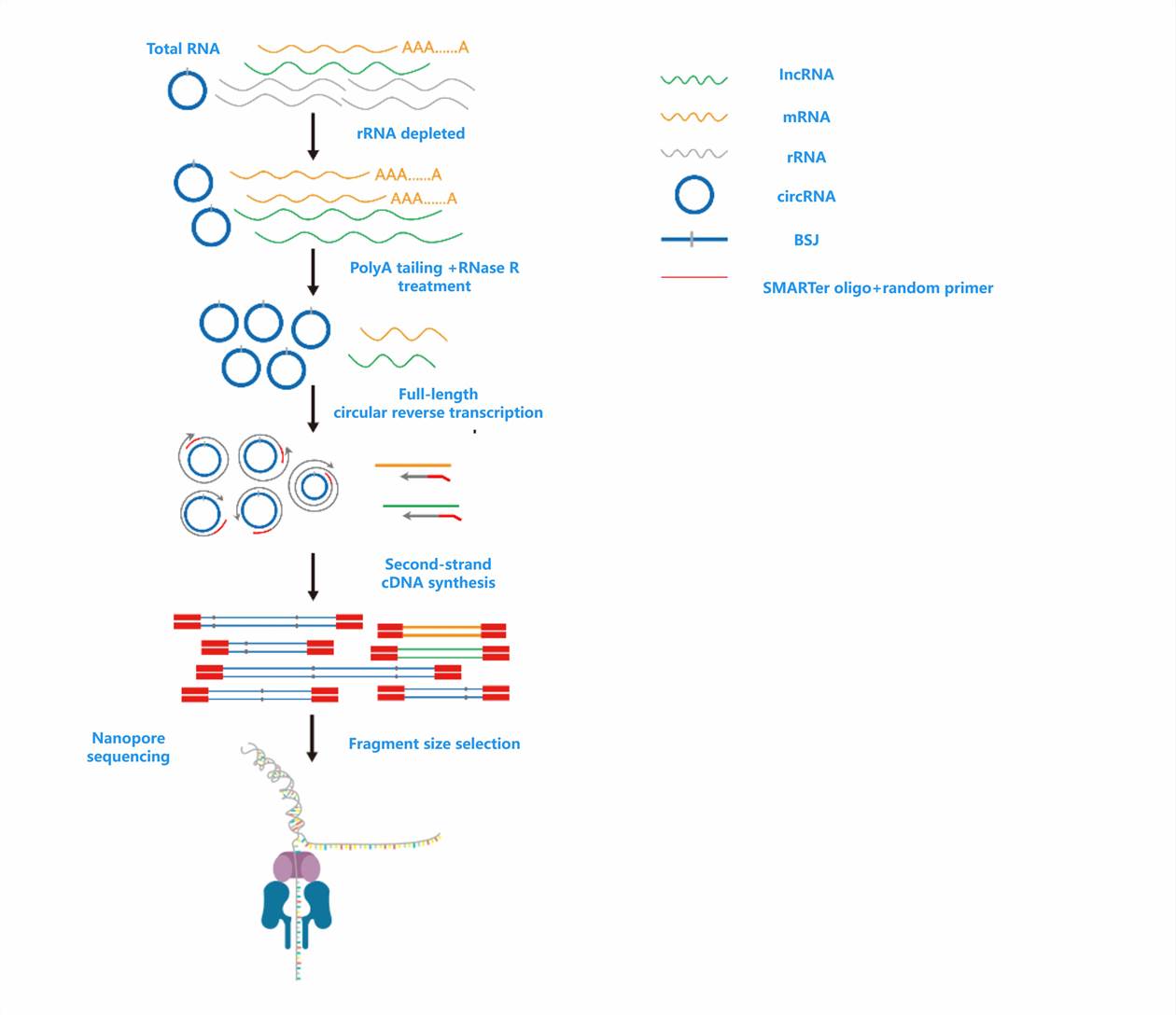
CD Genomics, leveraging the substantial advantages of long-read sequencing technology, effectively overcomes the limitations associated with short-read sequencing methods inherent in NGS. Recently, CD Genomics introduced a novel circRNA sequencing service grounded in nanopore sequencing technology, specifically targeting the precise identification and quantification of full-length circRNA sequences. This service employs random primers to perform rolling circle reverse transcription amplification on enriched circRNA, subsequently utilizing nanopore sequencing technology to sequence the entire length of circRNA directly. Additionally, through the integration of specific algorithms, this service facilitates the accurate evaluation of circRNA expression levels and the precise identification of full-length variants of transcriptional sequences.
circRNA Short-Read Sequencing VS Full-Length Sequencing
Read Length and Fragmentation: Traditional SRS platforms, such as Illumina, generate reads typically between 150-250 base pairs. While this is sufficient for many genomic applications, it poses significant challenges for capturing the full length of circRNAs, which often span 300-500 base pairs or longer. The short reads necessitate a complex and error-prone assembly process to reconstruct the complete circRNA sequences. In contrast, full-length sequencing technologies, particularly those utilizing nanopore sequencing, can produce reads that encompass the entire circRNA molecule, ensuring a more accurate and comprehensive capture of these structures.
Assembly Complexity: The process of assembling full-length circRNAs from short fragments is computationally intensive and prone to errors. This complexity arises because the assembly algorithms must piece together numerous short reads, which can lead to misassemblies or gaps in the reconstructed sequences. Full-length sequencing technologies bypass this issue by directly providing long reads that do not require extensive assembly, thus reducing errors and providing a more reliable representation of circRNA sequences.
BSJs: The identification of circRNAs relies heavily on detecting BSJs, where the 3' end of one exon is spliced to the 5' end of an upstream exon, forming a covalently closed loop. Short-read sequencing platforms often miss these crucial junctions due to their limited read length and the need for computational assembly. In contrast, full-length sequencing technologies like nanopore sequencing excel at detecting BSJs because the longer reads are more likely to span the entire circRNA, including the junctions. This capability leads to improved sensitivity and specificity in circRNA identification.
Expression Quantification: Accurate quantification of circRNAs is essential for understanding their biological roles and regulatory mechanisms. Short-read sequencing often introduces biases during PCR amplification and library preparation, which can distort the true abundance of circRNAs. Full-length sequencing technologies mitigate these issues by providing direct, long reads that do not require amplification, leading to more accurate quantification. This improvement is particularly important for low-abundance circRNAs, which are more susceptible to quantification errors in short-read sequencing.
Exon Usage and Intron Retention: CircRNAs can exhibit complex splicing patterns, including differential exon usage and intron retention, which are difficult to resolve with short-read sequencing. Full-length sequencing provides a clear advantage in this area by capturing entire circRNA molecules in single reads, allowing for direct observation of these splicing events. This detailed structural information is crucial for understanding the diverse functional roles of circRNAs and their involvement in gene regulation.
Non-Canonical Splicing Events: Beyond standard exon-exon junctions, circRNAs can include non-canonical splicing events, such as retained introns or non-GT/AG splice sites. These events are often missed or inaccurately represented in short-read sequencing due to the limitations of read length and assembly algorithms. Full-length sequencing offers a comprehensive view of these non-canonical events, providing deeper insights into the complexity of circRNA biogenesis and function.
circRNA Full-Length Sequencing Principle
CD Genomics employs nanopore sequencing for full-length circRNA identification and quantification. This technology leverages the unique properties of nanopores to sequence long RNA molecules in real-time, providing several key benefits:
Rolling Circle Amplification: CircRNAs are enriched using RNase R treatment to remove linear RNAs, followed by rolling circle amplification to generate long concatemers of the circRNA sequences. This process ensures that the circRNAs are sufficiently amplified for sequencing.
Nanopore sequencing: The amplified circRNA molecules pass through nanopores, generating electrical signals that are decoded into nucleotide sequences. This process allows for the capture of full-length circRNA sequences, providing a complete and accurate representation of the RNA molecule.
circRNA Full-Length Sequencing Workflow
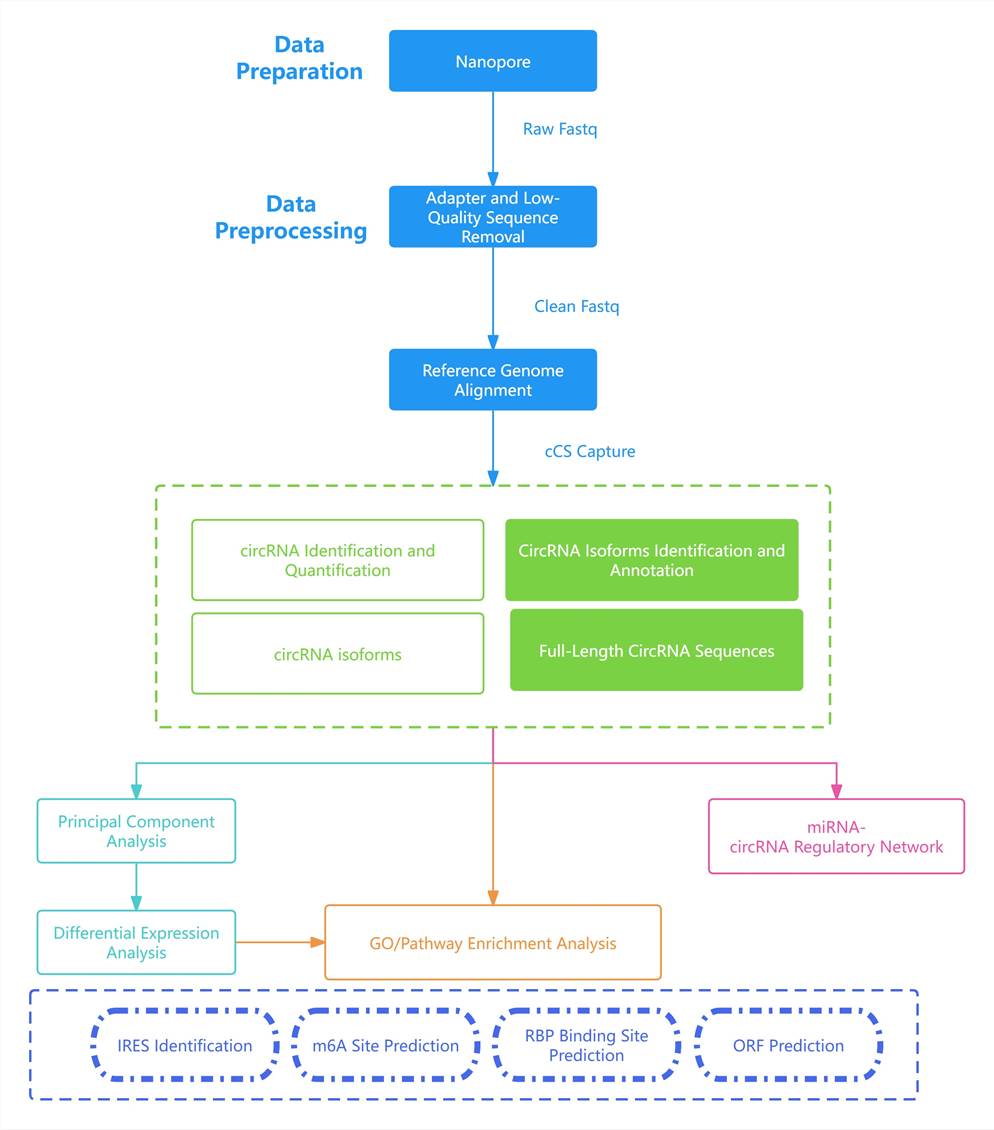
circRNA Full-Length Sequencing Bioinformatics Analysis
Advantages of circRNA Full-Length Sequencing Service
Enhanced Detection Efficiency: Over 20-fold improvement in circRNA read detection compared to SRS.
Higher Sensitivity: Capable of identifying low-abundance and non-classical circRNAs.
Accurate Detection of Alternative Splicing Events: Full-length RNA capture without the need for assembly, revealing complex circRNA structures.
Low RNA Input Requirement: Only 1 μg of total RNA needed, compared to 2 μg for SRS.
Efficient Workflow: The optimized protocols and advanced bioinformatics pipeline ensure a streamlined workflow from sample preparation to data analysis, delivering high-quality results.
Sample Requirements
Quality and Quantity of RNA Samples
| Parameter | Requirement | Notes |
| RNA Integrity Number (RIN) | ≥ 7 | Ensures high-quality RNA with minimal degradation. |
| Total RNA Amount | Minimum of 1 µg | Although 20 µg is ideal for maximizing circRNA detection, 1 µg is the minimum required. |
| Purity (A260/A280 Ratio) | 1.8 - 2.0 | Indicates minimal contamination with proteins, ensuring the RNA sample is pure. |
| Purity (A260/A230 Ratio) | ≥ 2.0 | Indicates minimal contamination with other organic compounds or salts. |
RNA Sample Preparation
| Step | Details | Purpose |
| DNase Treatment | Perform DNase I treatment to remove genomic DNA. | Eliminates DNA contamination which can interfere with accurate RNA sequencing. |
| RNA Extraction Method | Use column-based or phenol-chloroform extraction methods. | Ensures high yield and purity of RNA, minimizing the risk of residual contaminants. |
| rRNA Depletion | Employ rRNA depletion kits or strategies. | Removes ribosomal RNA, which can dominate the sequencing reads, allowing better circRNA detection. |
| Quality Check | Purity assessment. | Confirms the integrity and purity of the RNA sample before submission. |
Storage and Transport
| Parameter | Requirement | Notes |
| Storage Temperature | -80°C | Ensures the RNA remains stable and intact before sequencing. |
| Transport Conditions | Dry ice shipment recommended | Maintains RNA integrity during transport. |
| Packaging | Use RNase-free tubes and materials | Prevents RNase contamination which can degrade RNA samples. |
Special Considerations
| Factor | Recommendation | Notes |
| Sample Type | Prefer freshly isolated RNA, but preserved samples (e.g., frozen tissue, stabilized blood) are acceptable. | Fresh samples yield better integrity; however, preserved samples are suitable if handled properly. |
| CircRNA Enrichment | Consider using circRNA enrichment strategies if the circRNA content is expected to be low. | Enrichment can improve the detection sensitivity for low-abundance circRNAs. |
| Consultation | Contact CD Genomics for specific advice on sample preparation based on your experimental design. | Tailored recommendations can help optimize sample quality and sequencing results. |
References
- Liu et al., circFL-seq reveals full-length circular RNAs with rolling circular reverse transcription and nanopore sequencing. eLife, 2021.
- Xin et al. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nature communications, 2021.
- Zhang et al., Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long.Nature biotechnology, 2021.
- Rahimi et al., Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nature Communications, 2021.
- Qu et al., Circular RNA Vaccines against SARS-CoV-2 and Emerging Variants. Cell, 2022.
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment