While metagenomic sequencing has become a key tool, identifying genomic and functional pathways within the microbiome remains challenging in next-generation sequencing due to limitations associated with short (~300 bp) reads. Long-read sequencing, such as nanopore sequencing (ONT), is expected to circumvent these difficulties by providing longer reads (>3 kilobase pairs). Long read sequencing helps bridge inter-genomic repeats and produce better de novo assembly of genomes. Such hybrid metagenomic assemblies using short-read and long-read data have been used in human microbiome studies and metagenomic studies in various complex environments. Some cases are shown below.
OPERA-MS, a hybrid metagenomic assembler
The OPERA-MS process, which first uses MEGAHIT23, metaSPAdes24 or IDBA-UD25 for short sequence assembly, then uses long sequences to build the genomic framework, and then further bin out bacterial subspecies gene clusters. And then evaluate the assembly ability.
OPERA-MS, which combines metagenomic clustering with repeat-aware clustering, can accurately assemble complex bacterial communities. The authors found that OPERA-MS assembled more accurate base pairs than the long sequence assembler (Canu), more coherent assembly results than the short sequence assembler (MEGAHIT23, metaSPAdes24, IDBA-UD25), and lower error rates than the non-metagenomic hybrid assembler (hybridSPAdes). The process can also be assembled in the presence of multiple bacterial isoforms. The OPERA-MS can assemble high-quality genomes of sparse species (<1%) at long reads coverage of 9×, and nearly complete genomes at higher coverage.
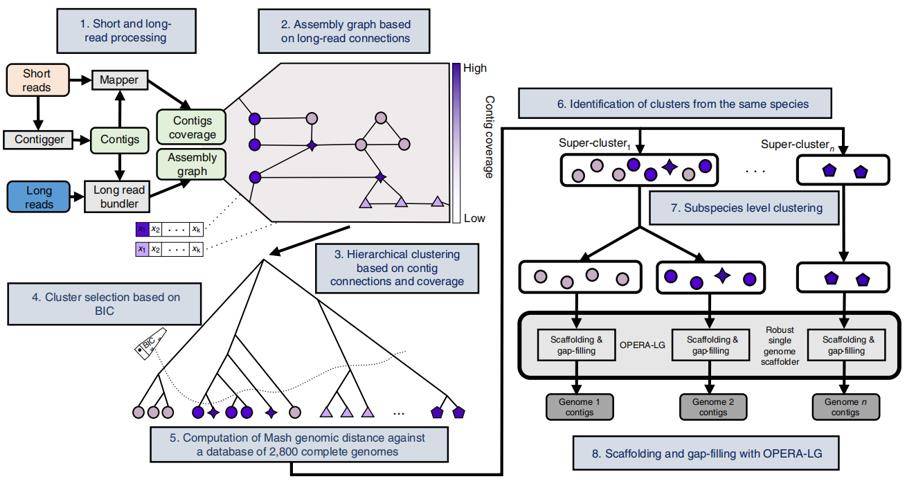 Workflow of OPERA-MS (Bertrand D et al. 2019)
Workflow of OPERA-MS (Bertrand D et al. 2019)
Hybrid assembly of metagenomic enables high-precision analysis of resistance genes and mobile elements
The analysis of microbiome composition has been achieved by high-throughput metagenomic sequencing. However, existing methods are not designed to assemble mixed sequences from short read length and long read length. The researchers used OPERA-MS hybrid metagenomic assembly software, which combines the assembly using repetition-aware clustering and precise scaffolding methods to achieve precise assembly of complex communities.
Researchers collected 197 clinical samples of intestinal colonized with carbapenem-resistant Enterobacteriaceae for long-read and short-read metagenomic sequencing. The intestinal metagenome of 28 antibiotic-treated patients was assembled using OPERA-MS and show that mixing data with nanopore long-read lengths yields more contiguous assemblies (a 200-fold improvement over short-read assemblies), including over 80 loop-forming plasmid or phage sequences and a new 263 kbp giant phage. High-quality hybrid assembly software allows for a fine-grained view of intestinal antibiotic resistance in human patients.
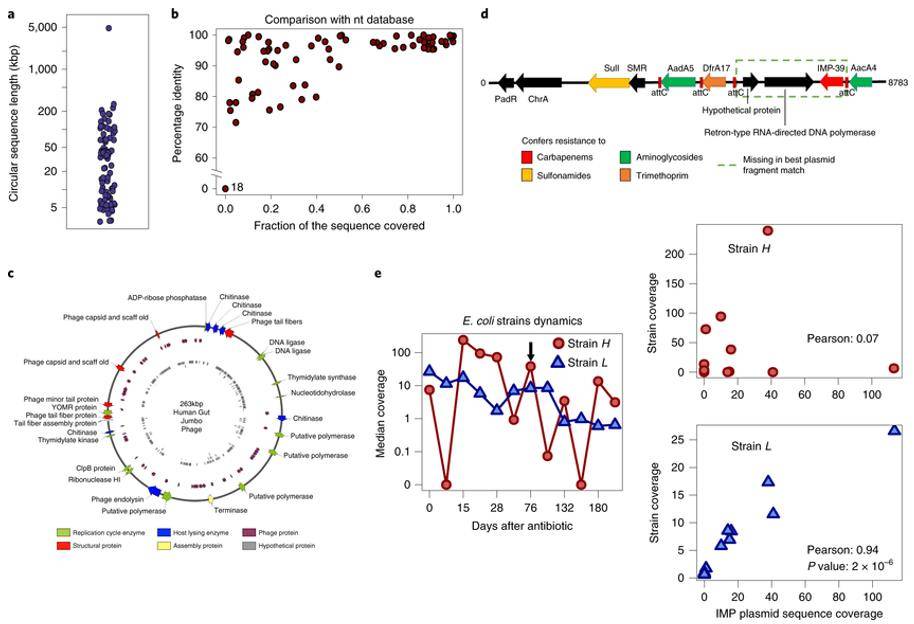 Mobile elements and association with host species in the human gut microbiome (Bertrand D et al. 2019)
Mobile elements and association with host species in the human gut microbiome (Bertrand D et al. 2019)
Hybrid metagenomic assembly links carbohydrate structure to function in the human gut microbiome
Microbial diversity within the microbiota and its interactions with host health and nutrition are now widely studied. An important role of the human gut microbiota is the metabolic breakdown of complex carbohydrates from plant and animal sources (e.g. legumes, seeds, tissues and cartilage). Short-chain fatty acids are the main product of carbohydrate fermentation by the intestinal microbiota. However, understanding the complexity of complex carbohydrate metabolism in the gut microbiota is a great challenge.
In this study, investigators used hybrid metagenomic assembly to assess species-level compositional changes in individual microbiota in the control model gut for six structurally distinct carbohydrates. Larger N50 and the longest overlapping clusters were found using hybrid assemblies compared to short-read assemblies. Five hundred and nine high-quality metagenome-assembled genomes (MAGs) belonging to 10 bacterial classes and 28 bacterial families were identified. Bacterial species identified as carrying starch-binding module genes showed a substantial increase in response to starch. Using the application of hybrid metagenomics, several uncultured species with functional potential to degrade starch substrates can be identified and can be used for future studies.
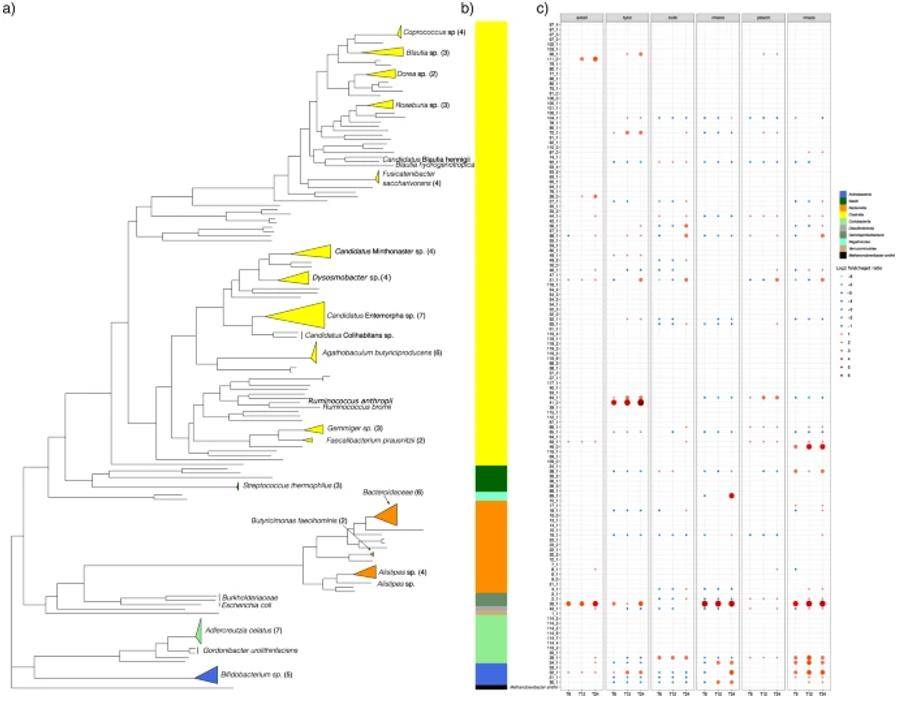 The phylogenetic tree was constructed from concatenated protein sequences using PhyloPhlAn and illustrated using ggtree (Ravi A et al. 2022)
The phylogenetic tree was constructed from concatenated protein sequences using PhyloPhlAn and illustrated using ggtree (Ravi A et al. 2022)
Summary
Deep long-read and short-read metagenomic sequencing and hybrid assembly have great potential for studying the human gut microbiota. Hybrid assembly of metagenomes results not only in high base accuracy, but also in an order of magnitude improvement in the splicing length of short-read sequencing data, thus allowing the assembly of genomes that are closer to complete, including high-quality genomes that can yield bacterial subspecies, rare microbes, plasmids and phages in complex samples.
References:
- Bertrand D, Shaw J, Kalathiyappan M, et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nature biotechnology, 2019, 37(8): 937-944.
- Ravi A, Troncoso-Rey P, Ahn-Jarvis J, et al. Hybrid metagenome assemblies link carbohydrate structure with function in the human gut microbiome. Communications Biology, 2022, 5(1): 1-13.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


