Most plants and animals have complex genomes with several features, such as large sizes, high heterozygosity, and polyploidy. Organisms are genetically diverse, and heterozygous genomic regions may be major contributors to phenotypic variation, and this complexity poses a challenge to genome assembly. The increase in the number of chromosome sets increases the total amount of DNA in the genome and increases genome complexity by adding alleles or other forms of genes. Although most sequences between paired chromosomes are identical, these differences provide the breadth of biological variation within species. The use of high-quality haplotype maps of the genome can provide a better understanding of the genetic history of a crop or animal, explore species domestication, and aid in species improvement research.
Haplotyping of polyploids requires, in principle, parental sequences, or if not available, at least their evolutionary ancestral species/near ancestral species sequences (for comparison to split different subgenomes), and to help mount them at a later stage.
The Strategies of Haplotype-Resolved Genome Assembly
Four main haploid genome assembly strategies are currently used by researchers.
The first strategy is the Trio binning (Illumina and PacBio sequencing) method that relies on parental sequences for efficient assembly. This method is simple and easy to implement, but prone to misclassification of reads when the parents are heterozygous.
The second strategy is the DipAsm (HiFi and Hi-C sequencing) method which does not rely on parental sequences and combines Hi-C data to produce chromosome-level haplotypes, but is prone to misclassification of highly heterozygous regions.
The third strategy is the Hifiasm method that effectively uses HiFi reads to generate high-quality haplotypes, which compared with DipAsm, not only maintains the advantage of not relying on the parents to assemble, but also reduces the dependence on Hi-C data, simplifies the process, achieves assembly and phasing in one click, and can integrate Hi-C data to help mount, and is gradually becoming the preferred method for high-quality assembly.
The fourth strategy is the polyploid genome assembly strategy, utilizing the PolyGembler or nPhase. The former requires the provision of lineage data and the latter requires the provision of reference genome sequences.
The diploid genome of marmosetreveals a specific evolutionary region of the Y chromosome
Callithrix jacchus is a small primate mammal and a common model animal for medical research. Using long-read and short-read sequencing data from marmoset families, the research team independently assembled two sets of high-quality haplotype genomes from each parent, which were published in Nature.
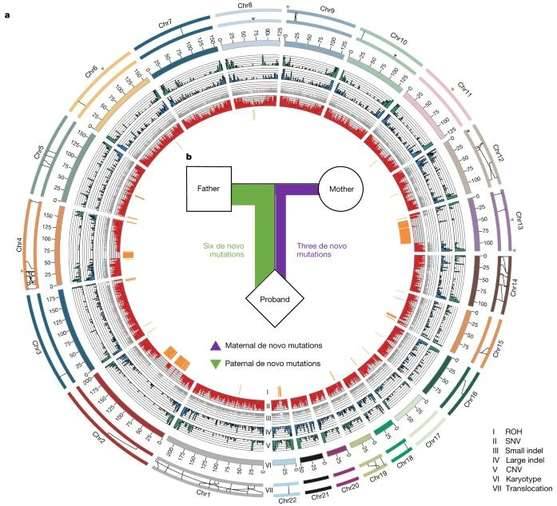 Heterozygosity landscape patterns between the two haploid marmoset genomes (Yang C et al., 2021)
Heterozygosity landscape patterns between the two haploid marmoset genomes (Yang C et al., 2021)
It was found that marmosets have an extra male-specific sequence on the Y chromosome compared to humans. Also, germline mutations from the parent were twice as high as those from the mother, possibly related to the different number of replicative cell divisions that occur during oocyte and sperm formation. The comparison of parental genome sequences refreshes the understanding of the differences in genetic information between parents and demonstrates the genetic basis of marmosets as a medical model species by analyzing growth and development-related genes. The related findings can be applied to studies in multiple directions such as neurodegenerative diseases, reproductive biology, and pharmacokinetic infectious diseases.
Apple haplotype genome study reveals origin and domestication history
Cornell University, in collaboration with USDA-ARS Plant Genetic Resources Research Center, has obtained high-quality genomic data through short-read and long-read sequencing of the cultivated apple (Malus domestica cv. Gala) and its major ancestral wild species, M. sieversii and M. sylvestris, high-quality haplotype genomes of apple were obtained.
Notably, haplotype-resolved genomes can help resolve the apple genome's origin and facilitate the study of allele-specific expression during species development. Several genes related to apple fruit development and quality were mined in this article, and the population evolution process of apples was revealed using population structure and population history analysis. This study provides precise and valuable genomic data for an in-depth study of apple domestication and genetic breeding.
Significance
The homologous chromosomes of diploid or polyploid species have high similarity, and the assembly process usually cannot distinguish the homologous chromosomes well due to the short-read length. But the long-read sequencing technology can help us identify the subtle differences between homologous chromosomes, and in combination with the assembly of other sequencing data, we can complete the haplotyping of diploids, identify the chromosomal differences from the parents, and further reveal the ancient origin and domestication process of the species.
Recommended Services
CD Genomics provides Whole Genome Sequencing based on Illumina and PacBio SMRT sequencing platforms, enabling rapid access to high-quality haplotype genomes, explaining more missing genetic power, and improving the accuracy of genome prediction.
References:
- Koren S, Rhie A, Walenz B P, et al. De novo assembly of haplotype-resolved genomes with trio binning. Nature biotechnology, 2018, 36(12): 1174-1182.
- Garg S, Fungtammasan A, Carroll A, et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nature biotechnology, 2021, 39(3): 309-312.
- Cheng H, Concepcion G T, Feng X, et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods, 2021, 18(2): 170-175.
- Zhou C, Olukolu B, Gemenet D C, et al. Assembly of whole-chromosome pseudomolecules for polyploid plant genomes using outbred mapping populations. Nature Genetics, 2020, 52(11): 1256-1264.
- Abou Saada O, Tsouris A, Eberlein C, et al. nPhase: an accurate and contiguous phasing method for polyploids. Genome biology, 2021, 22(1): 1-27.
- Yang C, Zhou Y, Marcus S, et al. Evolutionary and biomedical insights from a marmoset diploid genome assembly. Nature, 2021, 594(7862): 227-233.
- Sun X, Jiao C, Schwaninger H, et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nature genetics, 2020, 52(12): 1423-1432.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


