





We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Epigenetic profiling is essential for elucidating gene regulation and chromatin dynamics. Chromatin Immunoprecipitation sequencing (ChIP-seq) has long been the gold standard for mapping protein-DNA interactions and histone modifications. However, the recent advent of Cleavage Under Targets and Release Using Nuclease (Cut&Run) has introduced a powerful alternative technique, offering several advantages over traditional ChIP-seq methodologies.
The ChIP-seq methodology initiates with the cross-linking of proteins to DNA via formaldehyde, which stabilizes protein-DNA interactions. This crucial step preserves the native chromatin architecture, permitting subsequent isolation and analysis. Once cross-linking is complete, the chromatin is subjected to fragmentation through sonication or enzymatic digestion. Fragmentation is essential as it produces appropriately sized DNA fragments suitable for sequencing and enhances the efficiency of immunoprecipitation.
The cornerstone of the ChIP-seq technique is the immunoprecipitation stage, wherein chromatin fragments are incubated with antibodies specific to the protein of interest, be it transcription factors or histone modifications. These antibodies specifically recognize and bind to their target proteins, thereby enabling the selective enrichment of the corresponding protein-DNA complexes. Subsequently, the immunoprecipitated complexes undergo a series of washing steps to eliminate non-specifically bound chromatin and proteins. This washing process minimizes background noise and enhances the specificity of the results, ensuring greater accuracy in downstream analyses.
Following immunoprecipitation and subsequent washing, the reversal of cross-links is performed, leading to the extraction of DNA from the protein-DNA complexes. The resultant DNA fragments are then processed through high-throughput sequencing methodologies, yielding millions of short reads that are mapped to the genome. These sequencing reads are aligned to a reference genome, facilitating the identification of loci where the protein of interest exhibits binding affinity or where particular histone modifications are present.
ChIP-seq is extensively employed to delineate the binding sites of transcription factors across the genome. Through the identification of these binding loci, researchers can elucidate gene regulatory networks and gain insights into the mechanisms governing gene expression in various cellular contexts and under distinct conditions. For instance, ChIP-seq investigations have mapped the binding sites of essential transcription factors implicated in development, differentiation, and pathogenesis.
A notable example can be found in the study by Chen et al. (2008), which successfully mapped the binding sites of the transcription factors OCT4, SOX2, and NANOG in human embryonic stem cells. This study elucidated the roles of these transcription factors in maintaining pluripotency (Chen, X., et al., Cell, 2008).
Histone modifications are critical in the regulation of gene expression and the structuring of chromatin. ChIP-seq facilitates the detailed characterization of specific histone marks, including H3K4me3, H3K27ac, and H3K9me3. These marks are respectively correlated with active transcription, enhancer regions, and heterochromatin. Profiling these modifications permits researchers to elucidate chromatin states and gene expression patterns inherent to diverse biological processes and pathologies.
ChIP-seq has emerged as a pivotal methodology for examining epigenetic alterations implicated in various pathologies, including oncological and neurological disorders. By conducting comparative analyses of ChIP-seq profiles between normal and pathological tissues, researchers can discern aberrant protein-DNA interactions and atypical histone modifications contributing to disease etiology. These insights facilitate the identification of novel biomarkers and therapeutic targets.
For instance, the study conducted by Hnisz et al. (2013) leveraged ChIP-seq to identify super-enhancers within cancer cells. Super-enhancers are expansive conglomerates of regulatory elements that potentiate the expression of oncogenes, thereby underscoring potential therapeutic targets (Hnisz, D., et al., Cell, 2013).
ChIP-seq has significantly advanced the field of gene regulation by enabling a comprehensive analysis of protein-DNA interactions and chromatin modifications. This technique has facilitated the identification of novel regulatory elements, including enhancers and silencers, thereby enhancing the understanding of their roles in gene expression regulation. ChIP-seq investigations have elucidated the mechanisms underlying diverse biological processes, such as cellular differentiation, organismal development, and responses to environmental stimuli.
ChIP-seq has made substantial contributions to drug discovery and development by identifying potential drug targets and elucidating the mechanisms of drug action. This methodology has been employed to investigate the effects of small molecules on transcription factor binding and histone modifications, thereby providing insights into their therapeutic potential and mechanisms of action.
ChIP-seq data are frequently integrated with other genomic datasets, such as RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq), to yield a more comprehensive understanding of gene regulation and chromatin dynamics. This integrative methodology has significantly advanced the field of systems biology, enabling the modeling of complex gene regulatory networks and revealing interactions among various genomic elements.
Service you may intersted in
Cleavage Under Targets and Release Using Nuclease (Cut&Run) represents a powerful and innovative technique for the study of protein-DNA interactions and chromatin modifications, characterized by high precision and resolution. This section examines the principles, methodologies, applications, and advancements of Cut&Run, emphasizing its advantages and contributions to genomic research.
The Cut&Run method initiates with the targeted cleavage of chromatin using a specific nuclease. In contrast to traditional ChIP methods, which necessitate cross-linking and extensive washing steps, Cut&Run employs a high-affinity antibody conjugated to micrococcal nuclease (MNase). This approach facilitates the precise cleavage of DNA at the binding sites of the protein of interest, followed by the subsequent release of these DNA fragments from the chromatin.
Upon the execution of targeted cleavage by MNase, the DNA fragments linked to the protein of interest are liberated. This procedure exhibits a high degree of specificity, attributed to the exact binding affinity of the antibody to its designated protein. Subsequent to the release, the DNA fragments undergo purification, obviating the necessity for extensive washing protocols and thereby diminishing background interference. Consequently, this methodology yields a more refined and precise depiction of protein-DNA interactions in comparison to conventional ChIP-seq.
The purified DNA fragments are subsequently analyzed through high-throughput sequencing methodologies. The resultant sequencing reads are aligned to a reference genome, facilitating the identification of exact protein binding sites and chromatin modifications. The elevated resolution afforded by the Cut&Run technique enables the detection of fine-scale chromatin features and the accurate mapping of protein-DNA interactions.
The CUT&RUN technique offers a high-resolution perspective on protein-DNA interactions, which permits researchers to map binding sites with enhanced precision relative to conventional methodologies. This elevated resolution proves especially advantageous for examining transcription factor binding sites and other regulatory elements at a more refined scale. The superior accuracy provided by CUT&RUN augments the comprehension of gene regulation mechanisms and chromatin dynamics.
The CUT&RUN methodology can be utilized to examine specific histone modifications. By targeting histone marks indicative of various chromatin states, researchers can elucidate the epigenetic landscape of the genome. This technique is particularly valuable for investigating alterations in histone modifications associated with development, cellular differentiation, and pathological processes.
The precision afforded by the CUT&RUN technique facilitates the investigation of chromatin dynamics and structural alterations at high resolution. This methodology enables researchers to examine the influence of chromatin accessibility and structure on gene expression and cellular processes. Consequently, this application elucidates the mechanisms underlying chromatin remodeling and its effects on genomic function.
Cut&Run offers enhanced sensitivity and specificity relative to traditional chromatin assays. The utilization of high-affinity antibodies combined with targeted nuclease cleavage significantly reduces background noise, thereby improving the accuracy of the results. This increased sensitivity facilitates the detection of low-abundance proteins and subtle alterations in chromatin structure.
Cut&Run has significantly advanced disease research by elucidating the role of protein-DNA interactions and chromatin modifications in various pathologies. By comparing Cut&Run profiles between healthy and diseased tissues, researchers can identify aberrant interactions and modifications that contribute to disease etiology. These insights facilitate the discovery of novel biomarkers and therapeutic targets.
Data obtained from (Cut&Run is frequently integrated with other genomic techniques, such as RNA-seq and assay for ATAC-seq, to provide a comprehensive perspective on gene regulation and chromatin dynamics. This integrative approach enhances the understanding of the interactions among various genomic elements and their impact on gene expression and cellular function.
Cut&Run and ChIP-seq are both prominent techniques used to study protein-DNA interactions and chromatin modifications. Despite their shared goal of elucidating these interactions, they differ significantly in their methodologies, advantages, and applications.
| Comparison Aspect | Cut&Run | ChIP-seq |
| Chromatin Preparation | ||
| Chromatin Preparation | Begins with intact nuclei or chromatin without cross-linking. Allows for more specific and less biased targeting of protein-DNA interactions. | Requires cross-linking of proteins to DNA using formaldehyde, followed by chromatin fragmentation via sonication or enzymatic digestion. Cross-linking can introduce artifacts or non-specific interactions. |
| Nuclease Cleavage / Immunoprecipitation | Utilizes a micrococcal nuclease (MNase) coupled with a specific antibody to cleave DNA at protein binding sites, releasing only DNA fragments directly associated with the protein. | Proteins cross-linked to DNA are immunoprecipitated using specific antibodies. This is followed by extensive washing to remove non-specific material, which can sometimes result in loss of specific interactions. |
| Specificity and Resolution | ||
| Specificity | High specificity with reduced background noise due to targeted MNase cleavage. Absence of cross-linking and precise nuclease digestion ensure only relevant DNA fragments are captured. | Lower specificity due to reliance on cross-linking and extensive washing, which can sometimes fail to remove contaminants completely, affecting data specificity. |
| Resolution | High-resolution data, often better than ChIP-seq, capturing finer details of protein-DNA interactions for precise detection of binding sites and chromatin modifications. | Lower resolution due to broader chromatin fragmentation and cross-linking effects. The resolution is influenced by the size of chromatin fragments and extent of cross-linking. |
| Sensitivity and Background Noise | ||
| Sensitivity | Highly sensitive, requiring fewer starting materials. Capable of detecting low-abundance proteins and subtle chromatin modifications. | Requires larger amounts of starting material and may be less sensitive to low-abundance proteins. Sensitivity varies based on antibody quality and efficiency of chromatin immunoprecipitation. |
| Background Noise | Lower background noise due to high specificity, resulting in cleaner data with fewer non-specific interactions. | Higher background noise despite extensive washing, caused by non-specific interactions and incomplete removal of contaminants. |
| Data Interpretation | ||
| Data Interpretation | High resolution and specificity enable more accurate interpretation of protein-DNA interactions and chromatin modifications. Cleaner data allows precise mapping of binding sites and epigenetic marks. | Valuable for studying protein-DNA interactions; however, lower resolution and higher background noise can complicate interpretation. Broader chromatin fragments may obscure fine details of binding sites and modifications. |
| Applications | Particularly useful for studying specific transcription factor binding, histone modifications, and detailed chromatin features. | Widely used for studying global chromatin states, large-scale protein-DNA interactions, and genome-wide histone modifications. |
| Cost and Efficiency | ||
| Cost | More cost-effective due to lower material requirements and fewer processing steps. Reduced need for extensive washing and cross-linking further contributes to cost savings. | More expensive due to the requirement for large amounts of starting material, extensive washing, and additional steps such as cross-linking and fragmentation. |
| Efficiency | More efficient in terms of time and resources, providing high-quality data with minimal background noise and fewer experimental steps. | Less efficient in terms of time and resources, requiring extensive processing to obtain high-quality data. |
Cut&Run and ChIP-seq are both valuable techniques for studying protein-DNA interactions and chromatin modifications, each with its own set of advantages and limitations. Cut&Run offers higher resolution, specificity, and sensitivity, making it particularly suited for detailed studies of protein-DNA interactions and chromatin features. In contrast, ChIP-seq provides broader genomic coverage and is well-established for large-scale studies of chromatin states and global protein-DNA interactions. The choice between Cut&Run and ChIP-seq depends on the specific research goals, available resources, and desired resolution.
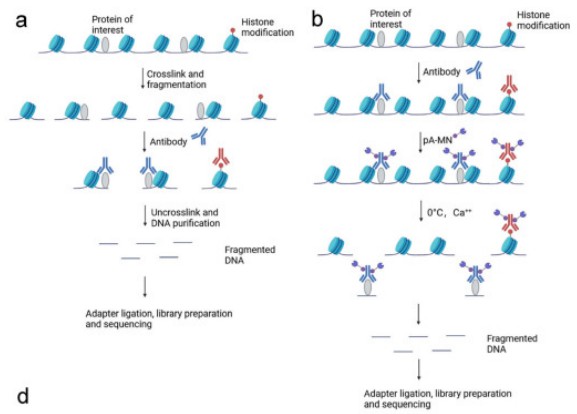 Figure 1. Schematic diagram of ChIP-seq, CUT&RUN, CUT&Tag, and DAP-seq. (a) ChIP-seq; (b) CUT&RUN (Mengyuan Wang et al,. 2023)
Figure 1. Schematic diagram of ChIP-seq, CUT&RUN, CUT&Tag, and DAP-seq. (a) ChIP-seq; (b) CUT&RUN (Mengyuan Wang et al,. 2023)
The accuracy of Cut&Run and ChIP-seq largely hinges on the specificity and affinity of the antibodies used. Antibodies must be rigorously validated to ensure they specifically bind to the target protein and not to non-specific sites. For example, in ChIP-seq studies, the choice of antibody significantly impacts the quality and reproducibility of the data. A well-validated antibody can distinguish between closely related histone modifications, which is crucial for precise chromatin mapping.
A study by Landt et al. (2012) emphasizes the importance of antibody validation in ChIP-seq. They developed and evaluated a comprehensive set of antibodies against various histone modifications and transcription factors, demonstrating that high-quality antibodies are critical for generating reliable ChIP-seq data. Their findings highlight the need for thorough validation to minimize background noise and improve data accuracy (Landt et al., 2012).
Optimizing experimental conditions, such as antibody concentration and incubation times, is crucial for achieving maximum enrichment and minimizing non-specific binding. For Cut&Run, optimization involves adjusting the concentration of the pA-MNase fusion protein and the incubation times to improve signal-to-noise ratios.
In their work, Skene et al. (2018) provided detailed protocols for optimizing Cut&Run conditions. They demonstrated how varying the concentration of MNase and antibody can significantly affect data quality, emphasizing the need for careful optimization to enhance the precision and reproducibility of results (Skene et al., 2018).
Both Cut&Run and ChIP-seq rely on high-throughput sequencing technologies to generate comprehensive datasets. The choice of sequencing platform impacts data quality and resolution. Platforms like Illumina and Oxford Nanopore offer different advantages, with Illumina providing high accuracy and read depth, while Oxford Nanopore offers longer read lengths that can be beneficial for analyzing complex chromatin landscapes.
Mikkelsen et al. (2007) utilized high-throughput sequencing to map histone modifications across the genome, illustrating the power of advanced sequencing technologies in generating detailed chromatin profiles. Their study underscores the importance of using state-of-the-art sequencing platforms to achieve high-resolution and comprehensive data (Mikkelsen et al., 2007).
Advanced bioinformatics tools are essential for interpreting the complex data generated by Cut&Run and ChIP-seq. Peak calling, motif analysis, and integration with other genomic datasets are critical steps in extracting meaningful biological insights. Tools such as MACS for peak calling and HOMER for motif analysis are widely used in the field.
Zhang et al. (2008) provided an overview of bioinformatics tools for ChIP-seq data analysis, including peak calling and motif discovery. Their study highlighted how these tools facilitate the interpretation of high-throughput sequencing data and enhance the understanding of chromatin dynamics (Zhang et al., 2008).
CUT&RUN and ChIP-seq are pivotal techniques for the investigation of protein-DNA interactions and chromatin modifications. Although ChIP-seq has been the prevailing method for an extended period, CUT&RUN presents several advantages, including enhanced sensitivity, reduced cellular input requirements, and minimized background noise. Both methodologies are indispensable for advancing research in epigenetics, elucidating disease mechanisms, and facilitating therapeutic development.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.