





We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Transcription factors (TFs) binding to specific locations in the genome influence dynamic changes in transcription levels. Disruption of TF binding sites (TFBS) correlates with phenotypic diversity and disease states, including agriculturally important adaptive traits and various diseases like cancer. Characterizing the genome-wide binding spectrum is crucial for exploring these mechanisms. DNA affinity purification sequencing (DAP-seq) is a high-throughput genomic sequencing technique allowing researchers to study TF-DNA binding sites in vitro while preserving 5-methylcytosine in genomic DNA, investigating the impact of DNA methylation on TF binding sensitivity. DAP-seq combines in vitro expressed affinity-tagged TFs with high-throughput sequencing of genomic DNA libraries, enabling the generation of genome-wide binding site maps reflective of both local sequence context and DNA methylation status. This is vital for a deeper understanding of complex transcriptional regulatory networks and their impact on organismal growth, development, and environmental adaptability.
CD Genomics offers comprehensive and accurate DAP-seq technology services, with experienced expert teams performing quality control at each step of the process to ensure result accuracy.
Service you may intersted in
DAP-seq technology can be used for the identification of individual transcription factor binding sites and target genes. In tomato, the TF BEL1-LIKE HOMEODOMAIN11 (SlBEL11) plays a crucial regulatory role in flavonoid biosynthesis, yet its regulatory mechanism remains unclear. Researchers conducted DAP-seq analysis and found that SlBEL11 does not directly bind to the promoter regions of flavonoid biosynthesis genes; instead, it binds to the promoter region of the SlMYB111 TF gene. SlMYB111 is a key regulatory factor in flavonoid biosynthesis, directly activating the expression of core flavonoid biosynthesis genes such as SlCHS1, SlCHI, SlF3H, and SlFLS. The application of DAP-seq revealed that SlBEL11 indirectly influences flavonoid biosynthesis by regulating the expression of SlMYB111, thereby modulating auxin output and distribution, ultimately affecting the abscission process of tomato fruits. This discovery holds significant implications for understanding plant internal regulatory mechanisms and improving crop quality and yield through molecular means. By identifying the direct targets of SlBEL11, researchers can gain deeper insights into its roles in plant development and response to environmental stimuli, offering potential molecular targets for future crop improvement endeavors.
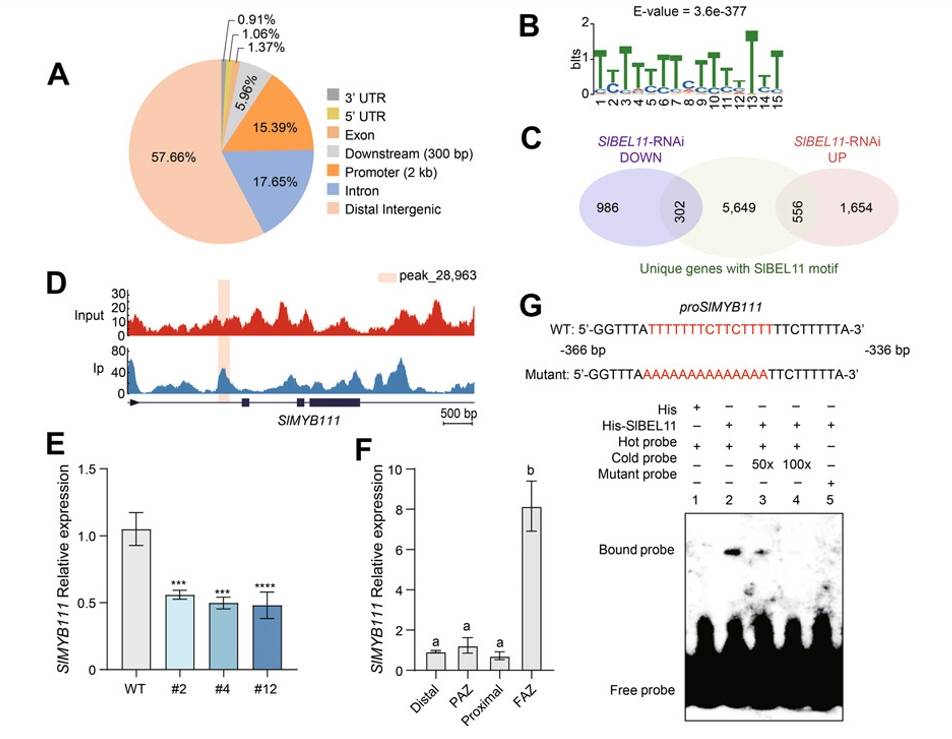 Solanum lycopersicum SlBEL11 directly promotes SlMYB111 expression in the tomato fruit abscission zone (FAZ)(Dong et al., 2024)
Solanum lycopersicum SlBEL11 directly promotes SlMYB111 expression in the tomato fruit abscission zone (FAZ)(Dong et al., 2024)
Furthermore, researchers have utilized DAP-seq technology to identify the binding motifs and target genes of the TF ONAC083 in rice. Further investigation revealed that OsNAC083 negatively regulates rice resistance to blast disease by binding to the ACGCAA element and influencing the transcription of OsRFPH2-6. In maize, a binding motif and target genes of an AP2/ERF TF have been identified. It has been elucidated that ZmEREB57 regulates maize OPDA synthesis through two distinct signaling pathways to enhance salt tolerance, providing valuable genetic resources for the breeding of salt-tolerant plant varieties. In poplar, the binding motif and target genes of the TF PtoWRKY68 have been identified. It has been revealed that allelic variation in the PtoWRKY68 gene regulates the molecular mechanism of response to drought stress by modulating the ABA signaling pathway, laying the genetic foundation for the development of drought-resistant tree varieties using molecular breeding strategies.
The DAP-seq technique can be utilized to study the binding sites of individual TFs. However, many eukaryotic TFs regulate gene expression by forming homodimers, heterodimers, or heterotetramers. Therefore, researchers have enhanced the DAP-seq technique to investigate the DNA binding specificity of homodimers, heterodimers, or heterotetramers.
The MADS TFs, SEPALLATA3 (SEP3), and AGAMOUS (AG) are essential for flower organ development, with SEP3-AG forming heterodimers or heterotetramers that co-regulate flower organ development. However, the regulatory mechanisms underlying this process remain unclear. Researchers have developed an improved technique termed sequential DAP-seq (seq-DAP-seq) to investigate the binding patterns of SEP3 homodimers, SEP3-AG heterodimers, and SEP3Δtet-AG heterotetramers across the entire genome. Through seq-DAP-seq, researchers precisely identified the genomic binding sites of both homodimeric and heterodimeric complexes formed by SEP3 and AG TFs. Furthermore, seq-DAP-seq analysis revealed the DNA binding specificity of different MADS TF complexes, including the impact of heterotetramer formation on DNA binding. By comparing the binding patterns of tetrameric and dimeric complexes, seq-DAP-seq uncovered the cooperative effects of tetrameric TF complexes on DNA binding affinity, as well as preferences for specific site intervals. In conclusion, seq-DAP-seq emerges as a powerful technique capable of elucidating the binding patterns of specific TF complexes across the entire genome, providing new tools and insights for the study of plant development and evolution.
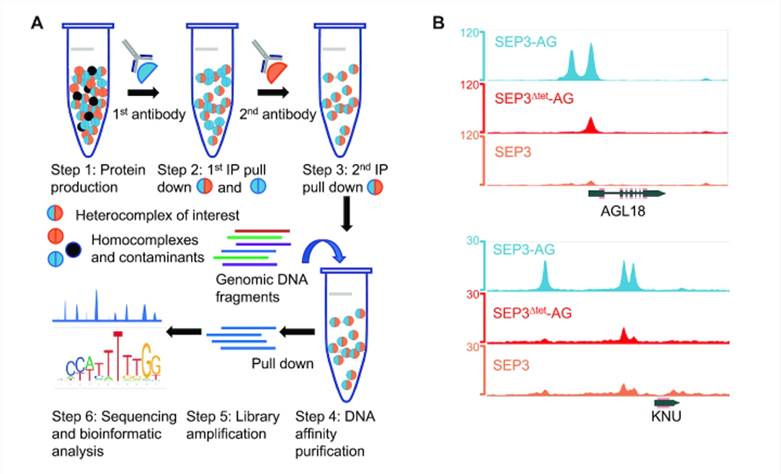 Seq-DAP-seq workflow and data analysis. (A) Schematic of the seq-DAP-seq protocol. (B) Integrated Genome Browser snapshot of SEP3–AG (blue), SEP3Δtet-AG (red) and SEP3 (orange) peaks from seq-DAP-seq showing complexes bind unique sites and common sites with varying intensity. (Lai et al., 2020)
Seq-DAP-seq workflow and data analysis. (A) Schematic of the seq-DAP-seq protocol. (B) Integrated Genome Browser snapshot of SEP3–AG (blue), SEP3Δtet-AG (red) and SEP3 (orange) peaks from seq-DAP-seq showing complexes bind unique sites and common sites with varying intensity. (Lai et al., 2020)
Plant BASIC LEUCINE ZIPPER (bZIP) TFs play crucial roles in various biological processes such as flower development, seed maturation, dormancy, and senescence by forming homodimers or heterodimers through their leucine zipper structures and subsequently binding to specific gene promoters via their basic regions to regulate the expression of downstream target genes. Researchers have developed an improved technique called Double DAP-seq (dDAP-seq) to reveal the binding sites of interacting bZIP TFs on plant genomic DNA. This technique combines two different affinity tags (such as HaloTag and SBPTag), allowing for the co-expression and complex formation of two different TFs in vitro. Subsequently, specific affinity beads are used to capture these TF complexes along with their bound DNA fragments, and high-throughput sequencing is employed to determine the precise binding sites of these TFs in the genome.Through dDAP-seq technology, researchers can uncover the differences in DNA binding specificity between heterodimers and homodimers of TFs, which is crucial for understanding how plants adapt to different environmental and developmental conditions by finely regulating gene expression. Using dDAP-seq, researchers identified the binding sites of C/S1 heterodimers and homodimers in Arabidopsis and analyzed the functional significance of these binding sites, including the role of bZIP9 in ABA response and the role of bZIP53 heterodimer-specific binding in seed maturation. The study also found that the formation of heterodimers not only increases the quantity and complexity of TFs but also enhances their functional specificity. In summary, dDAP-seq technology provides a powerful tool for studying the DNA binding specificity and combinatorial gene regulation of TFs, revealing the diverse functions and regulatory mechanisms of C/S1 bZIP TFs in plant biology.
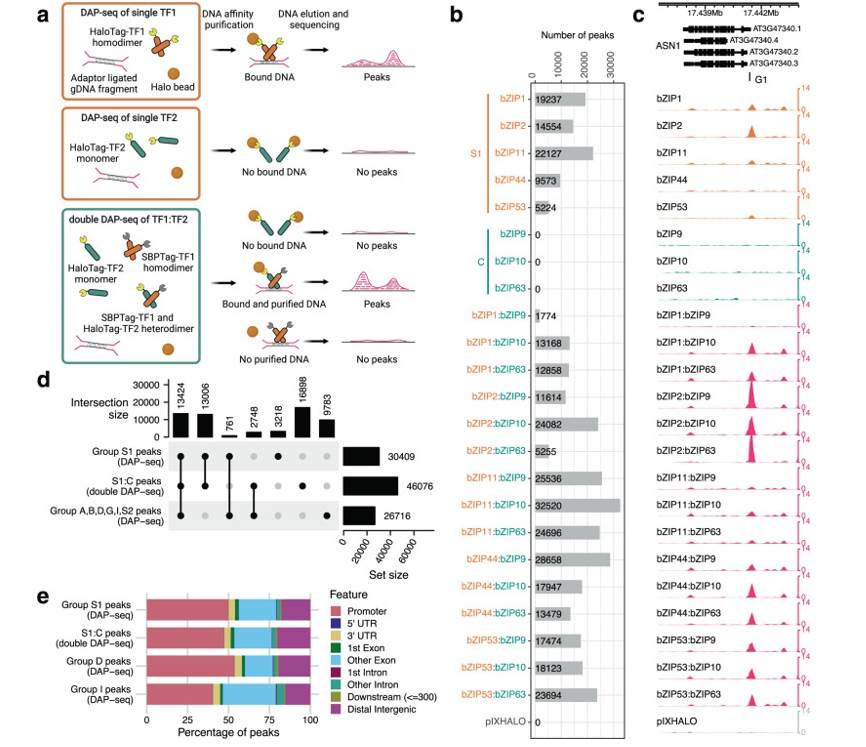 Systematic identification of bZIP C/S1 bZIP homodimer and heterodimer binding sites by DAP-seq and dDAP-seq. a. Schematic of DAP- and dDAP-seq; b. Number of peaks from all pairs of C/S1 bZIPs detected by DAP-seq and dDAP-seq; c. DAP-seq and dDAP-seq binding profiles of C/S1 homodimers and heterodimers at the known target gene ASN1 (AT3G47340); d. Upset plot comparing the peak overlap between S1 homodimers, S1:C heterodimers and bZIPs from group A, B, D, G, I, and S2. The dot plot lists the possible combinations between the different bZIP groups; e. Distribution of binding sites relative to genomic features for the bZIP homodimers and heterodimers. (Li et al., 2023)
Systematic identification of bZIP C/S1 bZIP homodimer and heterodimer binding sites by DAP-seq and dDAP-seq. a. Schematic of DAP- and dDAP-seq; b. Number of peaks from all pairs of C/S1 bZIPs detected by DAP-seq and dDAP-seq; c. DAP-seq and dDAP-seq binding profiles of C/S1 homodimers and heterodimers at the known target gene ASN1 (AT3G47340); d. Upset plot comparing the peak overlap between S1 homodimers, S1:C heterodimers and bZIPs from group A, B, D, G, I, and S2. The dot plot lists the possible combinations between the different bZIP groups; e. Distribution of binding sites relative to genomic features for the bZIP homodimers and heterodimers. (Li et al., 2023)
The gene regulatory network, comprising TFs, epigenetic modifiers, and other regulatory elements, orchestrates a complex interplay crucial for gene expression regulation within organisms. Delving deeply into this network aids in unraveling the mechanisms and biological functions underpinning gene expression regulation, thereby advancing our understanding of the essence of life and the onset and progression of diseases. And DAP-seq technology contributes to the construction of gene regulatory networks.
Researchers leveraged DAP-seq technology to identify and analyze the TFs governing pigment gland formation in cotton, alongside their downstream genes. By subjecting Gossypium Pigment Gland Formation (GoPGF) to DAP-seq analysis, DNA sequences bound to it, potentially harboring cis-regulatory elements modulating pigment gland formation, were unveiled. Integration of DAP-seq data with single-cell RNA sequencing (scRNA-seq) data facilitated the construction of a TF regulatory network centered around GoPGF. This network encompasses various TFs, including AP2/ERF, bHLH, C2H2, GRAS, NAC, and WRKY, pivotal in pigment gland formation and development. Furthermore, DAP-seq technology aided in validating downstream target genes of GoPGF, further refining the potential gene targets involved in regulating pigment gland development. Through the combined analysis with scRNA-seq data, researchers proposed a model elucidating cotton pigment gland formation, encompassing developmental trajectories of pigment gland cells, TF regulatory networks, and functional validation of core TFs. In conclusion, DAP-seq technology plays a pivotal role in exploring gene regulatory networks, furnishing a potent toolset for unraveling the mechanisms of gene expression regulation and the structural intricacies of regulatory networks within organisms.
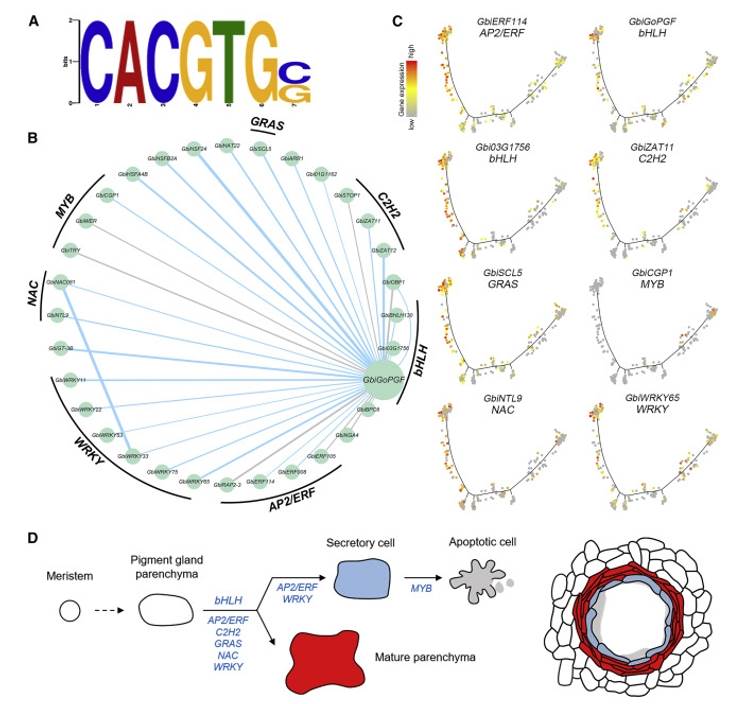 TF regulatory network of pigment gland cell development (Sun et al., 2023)
TF regulatory network of pigment gland cell development (Sun et al., 2023)
In summary, the DAP-seq technique ingeniously integrates in vitro protein expression with high-throughput sequencing technology, facilitating comprehensive identification and analysis of TF binding patterns across the entire genome. Simultaneously, integration with multi-omics analysis is crucial for gaining deeper insights into the intricate transcriptional regulatory networks and their pivotal roles in governing biological growth, development, and environmental adaptability.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.