Over the last two decades, the zebrafish has emerged as a widely embraced model organism due to its rapid embryonic development and the transparent nature of its externally fertilized embryos, rendering it exceptionally suitable for developmental research. Its compatibility with both forward and reverse genetic approaches has significantly facilitated gene discovery and the modeling of human diseases. In 2013, the Sanger Institute accomplished the sequencing of the zebrafish genome, encompassing a vast 1.5 million square meters. The zebrafish genome, which comprises approximately 1.4 Gb and encodes a minimum of 25,000 genes, is comparable in size to genomes of other vertebrates, and intriguingly, more than 70% of human genes exhibit homology with their zebrafish counterparts. However, the zebrafish genome presents a notable challenge akin to the human genome, mainly in the form of non-coding annotations.
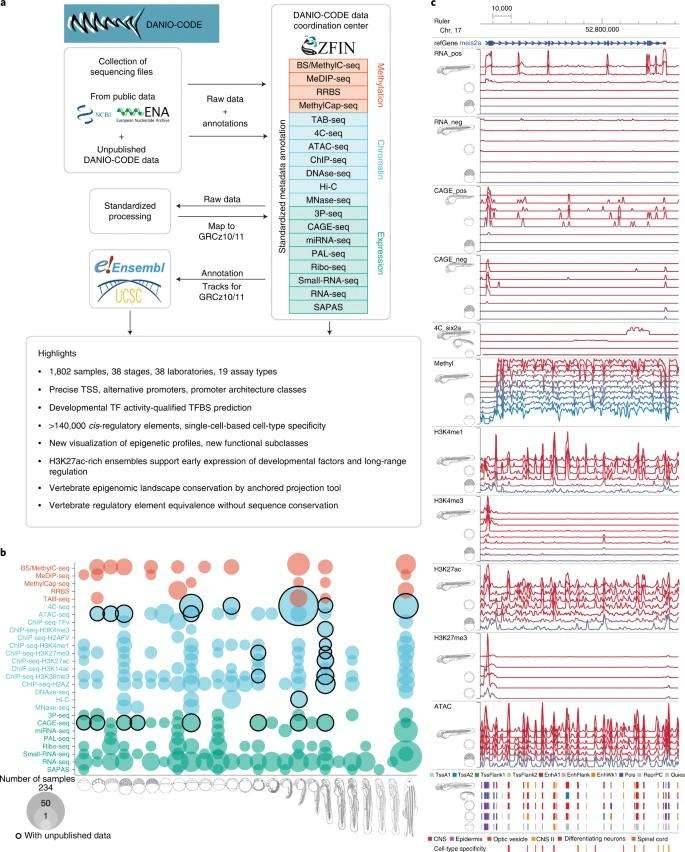
This research implemented a methodology akin to other collaborative projects such as ENCODE. Leveraging this strategy, the researchers re-examined a substantial body of previously published datasets, numbering almost 1,500, in addition to introducing over 350 new datasets. These datasets encompassed various high-throughput techniques, including ChIP-seq for profiling chromatin modifications associated with promoters and enhancers, ATAC-seq for identifying accessible regions in the genome, RNA-seq for the construction of gene models, Cap Analysis of Gene Expression (CAGE) for pinpointing 5' transcriptional endpoints, and Hi-C or 4C-seq for uncovering intrachromosomal interactions. Notably, this extensive collection of datasets spanned 15 different developmental stages as well as adult tissues, allowing for a dynamic assessment of genomic alterations during embryogenesis. The outcomes of this study are readily accessible and can be explored through the UCSC Genome Browser.
 Comprehensive collection and annotation of zebrafish developmental genomic data. (Baranasic et al., 2022)
Comprehensive collection and annotation of zebrafish developmental genomic data. (Baranasic et al., 2022)
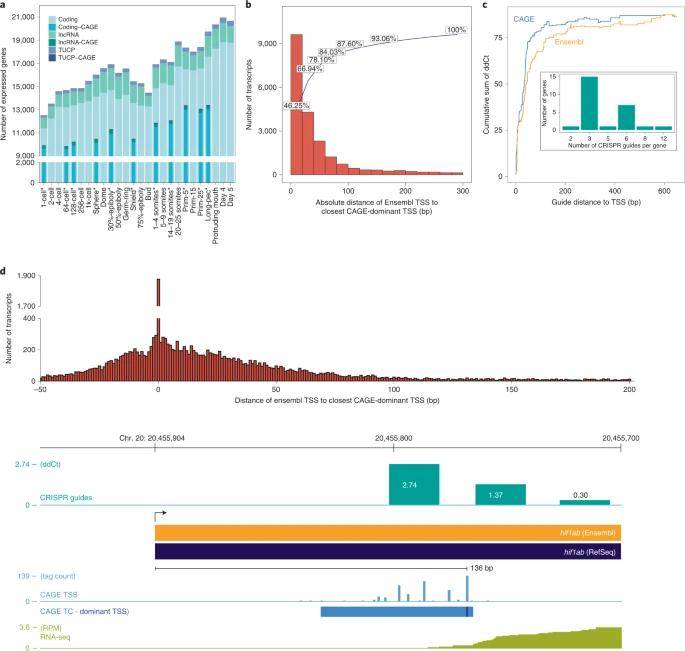
The authors leveraged their dataset to meticulously annotate non-coding elements within the zebrafish genome, with a primary focus on promoters and enhancers. To pinpoint promoter regions, they initiated the process by utilizing RNA-seq data to identify gene models. Subsequently, they incorporated CAGE reads, which specifically capture the 5' end of transcripts and correspond to the promoter region. This meticulous approach culminated in the precise determination of transcription start sites.
It's crucial to note that the dataset spans various embryonic stages, offering an intricate temporal perspective on how promoters evolve during development. To validate the accuracy of this "promoter set," the authors employed dCas9, an enzyme designed to incapacitate Cas9, thus preventing the binding of transcriptional activators at promoters and consequently reducing gene expression at selected promoter sites. Remarkably, directing dCas9 to the transcription start sites defined by CAGE data yielded more potent gene repression compared to sites defined by Ensembl annotations, underscoring the superior accuracy of the former in identifying active promoters. This invaluable resource empowers researchers with a robust foundation for conducting knockout studies.
In their quest to delve deeper into promoter characterization, the authors delved into chromatin modifications, accessibility dynamics across developmental time, and the assessment of sequence conservation. These comprehensive analyses unveiled a multitude of unique promoter structures characterized by dynamic activation patterns throughout embryonic development. While the biological significance of these diverse activation patterns remains a mystery, they serve as a promising point of departure for future hypothesis-driven experiments and investigations.
 Transcript categories and single-nucleotide resolution 5′ end verification during development. (Baranasic et al., 2022)
Transcript categories and single-nucleotide resolution 5′ end verification during development. (Baranasic et al., 2022)
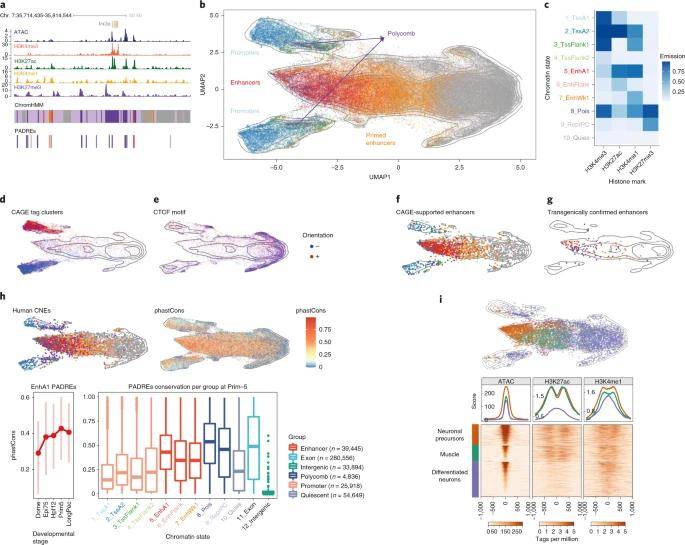
This study also offers an initial annotation of active enhancers within the zebrafish genome, achieved through the integration of accessibility and chromatin modification patterns. Their comprehensive analysis led to the identification of more than 100,000 elements exhibiting predicted enhancer activity. These elements can be further classified based on their dynamic activation patterns at various developmental stages. To confirm the functionality of these enhancers, the researchers assessed them through co-expression of nuclear cage enhancer RNAs and compared their findings with previously published reports.
Subsequently, the authors harnessed available single-cell ATAC-seq data to make predictive assessments of the cell-specific activity of approximately 40,000 enhancers. Many of these predictions found support in published reporter analyses. The study further delved into enhancer-promoter interactions by integrating data from intrachromosomal interactions using Hi-C and 4C-seq datasets. This analysis revealed a distinct genomic signature referred to as the H3K27ac sequence, sharing common characteristics with super-enhancers. Interestingly, the H3K27ac gene cluster appeared broader and more numerous than super-enhancers and was linked to the expression of early developmental genes before lineage specification.
Of notable significance, the authors introduced an innovative approach to compare genomes of distantly related species. This approach allowed them to identify co-lineage regulatory elements shared between mouse and zebrafish, underscoring the potential conservation of the H3K27ac sequence in vertebrate genome regulation. As a result, the exhaustive annotation of the zebrafish genome undertaken by the DANIO-CODE initiative holds the potential to facilitate the identification of unique, conserved, and developmentally relevant genomic regulatory features.
 Classification of developmental cis-regulatory elements. (Baranasic et al., 2022)
Classification of developmental cis-regulatory elements. (Baranasic et al., 2022)
Zebrafish, renowned as a model organism for developmental studies, has proven invaluable in the exploration of functional non-coding sequences crucial for both transcriptional and post-transcriptional regulation. Anticipating the next stages of research, it is expected that we will delve deeper into the functional characterization of the elements identified in our initial analyses. For instance, this may involve reporter assays, which can be readily and swiftly performed within live zebrafish embryos.
Furthermore, the inclusion of single-cell molecular techniques is imperative to refine our understanding. The preliminary DANIO-CODE investigations predominantly relied on whole embryos, introducing potential complexities in interpreting the current findings. Given the scarcity of zebrafish cell lines, the progression of technology in molecular analysis at a single-cell level and its subsequent application to zebrafish embryos will be pivotal in the forthcoming phases of DANIO-CODE.
Reference:
- Baranasic, Damir, et al. "Multiomic atlas with functional stratification and developmental dynamics of zebrafish cis-regulatory elements." Nature genetics 54.7 (2022): 1037-1050.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


