The clinical outcomes of SARS-CoV-2 exhibit substantial variation among individuals, with risk factors encompassing advanced age, male gender, comorbidities, and host genetics, as supported by extant research.
To elucidate the influence of natural selection and historical genome mixing on the immune response to SARS-CoV-2, a comprehensive study was conducted. The research team assembled a cohort of 220 individuals spanning diverse genders and ancestral backgrounds. These individuals underwent single-cell peripheral blood sequencing, encompassing infections by SARS-CoV-2 and influenza A virus, alongside matched control subjects. This yielded a robust dataset of one million high-quality single-cell transcriptomes, unveiling 22 distinct cell types in the bone marrow, as well as B cells, CD4+ T cells, CD8+ T cells, and natural killer (NK) cells within the immune profile. Subsequently, the study meticulously examined the distinct effects of genetic variation and natural selection on immune responses through an in-depth analysis of eQTL (expression quantitative trait loci) and natural selection dynamics. The findings from this pioneering research were published in Nature.
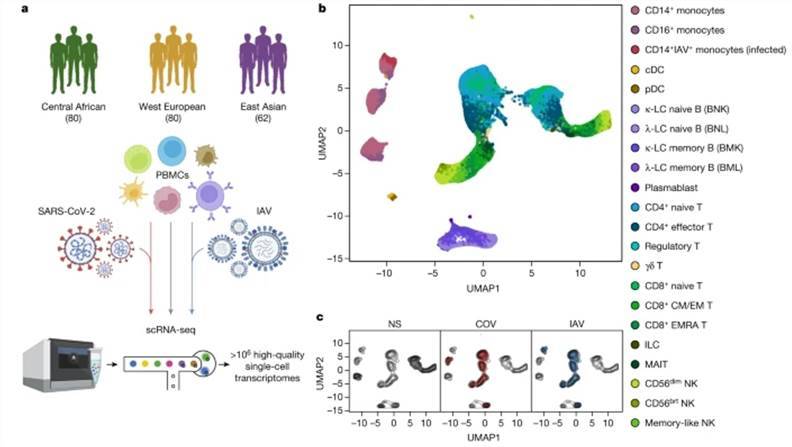 Population single-cell responses to SARS-CoV-2 and IAV. (Aquino et al., 2023)
Population single-cell responses to SARS-CoV-2 and IAV. (Aquino et al., 2023)
Cellular Heterogeneity in Latent Cytomegalovirus Infection Varies Across Different Genetic Backgrounds
Significant disparities in cellular composition were observed among distinct genetic background cohorts, with particular prominence in NK cells. Notably, a subset of memory-like NK cells accounted for 55.2% of the NK cell population in individuals of African descent, whereas the proportion was only 12.2% in Europeans. Additionally, African donors exhibited higher proportions of CD16+ monocytes and subpopulations of memory lymphocytes, including memory B cells, effector CD4+ T cells, and effector memory re-expressing CD45RA CD8+ T cells.
However, the driving force behind these changes in cellular fractions was not attributed to genetic factors but rather linked to latent cytomegalovirus (CMV) infection. The researchers verified the CMV serostatus (CMV+/-) of the samples and found that individuals of African descent were predominantly CMV+ (99%), in contrast to only 31% of Europeans. CMV+ status was correlated with a higher proportion of memory-like NK cells and CD8+ EMRA T cells in Europeans.
Gene expression patterns also exhibited variations among populations, with 898 and 652 genes displaying differential responses following SARS-CoV-2 and influenza A virus (IAV) stimulation, respectively. For instance, both IFN-responsive GBP7 and the gene encoding the macrophage inflammatory protein MIP-3 CCL23 were more significantly upregulated in Europeans. Notably, the CCL23 gene exhibited myeloid cell-specific upregulation in response to SARS-CoV-2 stimulation.
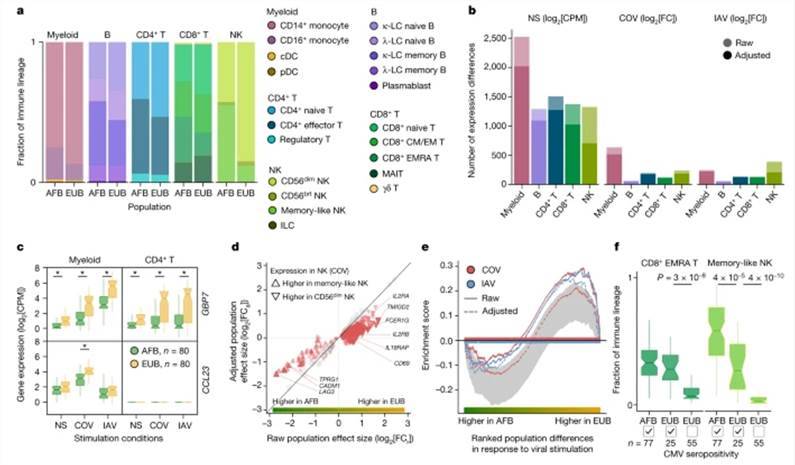 Cellular composition affects the transcriptional responses to viral stimuli. (Aquino et al., 2023)
Cellular composition affects the transcriptional responses to viral stimuli. (Aquino et al., 2023)
Natural Selection Exerts A Significant Influence on Variations in Population Immune Responses
A comprehensive analysis revealed the presence of 1,616 eQTLs (associated with 1,215 genes) and 180 reQTLs (related to 166 genes) identified using population branching statistics (PBS). This investigation aimed to uncover connections between (r)eQTLs and genome-wide localized adaptive signals, with 90 instances being specific to particular ancestral groups.
Among the genes associated with adaptive (r)eQTLs, researchers pinpointed key players in interferon-mediated antiviral immunity. In African populations, genes like DHX58 and TRIM14 were highlighted, while Europeans exhibited significance in ISG20, IFIT5, BST2, and IFITM2-3. East Asians, on the other hand, showed relevance in IFI44L and IFITM2.
The study uncovered rapid adaptation signals that targeted the same (IFITM2, IFIT5) or different (ISG20, IFITM3, TRIM14) eQTLs within highly differentiated genes. This suggests the presence of duplicate adaptations in response to interferon-mediated antiviral immunity. Of particular interest, 34% of genetically determined differentially expressed genes in African and European populations exhibited signals of rapid adaptation within the European genetic context.
Furthermore, by comparing these findings to ancient Neanderthal variants, researchers observed that ancient haplotypes were 1.4-1.5 times more likely to influence gene expression in the European ancestral state, both before and after exposure to SARS-CoV-2 or IAV stimulation. Notably, this enrichment was most pronounced in SARS-CoV-2-stimulated CD16+ monocytes from Europeans, indicating a preference for the retention of ancient haplotypes that modify myeloid responses in their genomes. This observation underscores the adaptive nature of Neanderthal regulatory alleles.
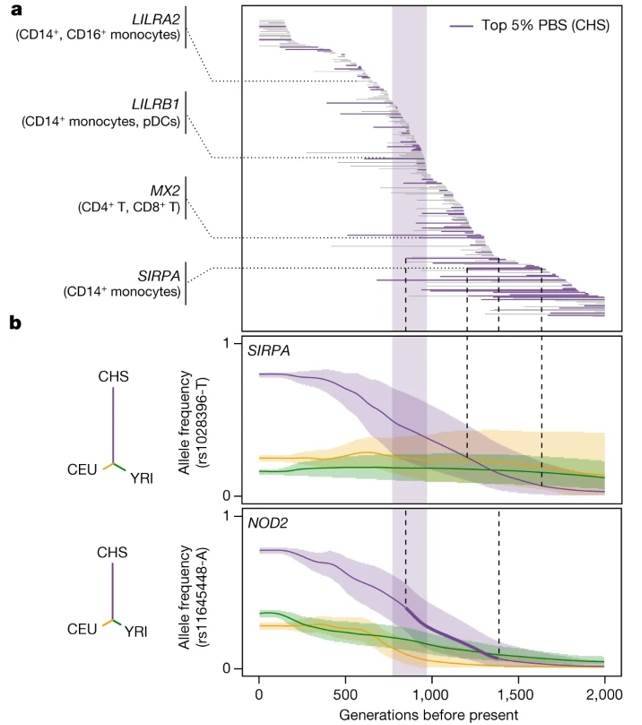 Natural selection effects on population differentiation of immune responses. (Aquino et al., 2023)
Natural selection effects on population differentiation of immune responses. (Aquino et al., 2023)
Differential Expression and Population-Specific Effects Potentially Driven by Genetic Factors
Utilizing eQTLs, our analysis revealed the identification of 9,150 eQTL loci associated with 5,198 distinct genes, of which 11% displayed ancestry-specific patterns. Among these, 812 eQTLs exhibited cell-type specificity, with 45% of them being predominantly detected in myeloid cells. Furthermore, we identified 1,505 viral-stimulated-response variants, known as reQTLs, linked to 1,213 genes. Notably, the genetic underpinnings of the myeloid response displayed viral specificity, with variations across cell populations.
Our investigation into inter-population differentially expressed genes and differential effect genes demonstrated that 56% and 60% of them, respectively, were associated with at least one eQTL. This suggests that the genes exhibiting the most significant population-based variation in differential expression and effects are likely influenced by genetic control, often linked to substantial effect-r loci. This highlights the presence of population differences in specific subsets of genes. For instance, between Africans and Europeans, we found that a single variant with strong population differentiation could explain 81-100% of the variation in GBP7 expression.
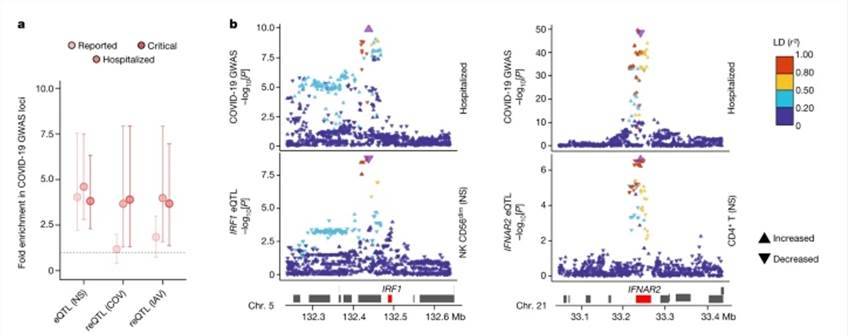 eQTLs and reQTLs contribute to COVID-19 risk. (Aquino et al., 2023)
eQTLs and reQTLs contribute to COVID-19 risk. (Aquino et al., 2023)
The article offers valuable insights into the factors influencing variations in immune responses among individuals and populations. It accomplishes this by leveraging single-cell data from diverse genetic backgrounds, shedding light on the environmental, genetic, and evolutionary drivers of these variations. This innovative application of the single-cell methodology to the realms of population genetics and immunity underscores its profound significance in capturing the entirety of diversity within these domains.
Reference:
-
Aquino, Yann, et al. "Dissecting human population variation in single-cell responses to SARS-CoV-2." Nature (2023): 1-9.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


