The ability of tumor cells to spread and metastasize to normal tissues presents a formidable challenge in the quest to cure cancer. Metabolic reprogramming, often exemplified by the Warburg effect, lies at the heart of this process. While normal cells predominantly engage in oxidative phosphorylation when oxygen is available and glycolysis in its absence, tumor cells opt for glycolysis even in the presence of oxygen. However, this metabolic preference doesn't preclude the existence of oxidative phosphorylation in tumor cells. Instead, tumor cells exhibit remarkable metabolic plasticity, enabling them to adapt to hostile environments, maintain metabolic stability, and, ultimately, thrive, metastasize, and persist.
Mitochondria serve as pivotal hubs for energy production, substance synthesis, and signal transduction within cells. Research indicates that the mitochondrial genome encodes 22 tRNAs critical for the 13 subunits of the respiratory chain complex. These tRNAs undergo over 90 modifications, and any deletion or mutation of the enzymes responsible for tRNA modification can disrupt protein synthesis efficiency and various cellular processes, leading to the development of numerous diseases.
A groundbreaking study conducted by researchers at the German Cancer Research Center, published in Nature, has shed light on the pivotal role of mitochondrial RNA modifications in shaping metabolic plasticity during metastasis. This study underscores that RNA-specific modifications play a central role in the metastatic process and offer a promising avenue for limiting the metastatic potential of tumors. Notably, modifications m5c and f5c at site 34 of mitochondrial genome tRNA, encoding methionine, emerge as key regulators of mitochondrial metabolism, furnishing the necessary energy to drive metabolic reprogramming.
Cytosine Modification in Mitochondrial tRNA-Met
In their investigation, the authors initiated their exploration of mitochondrial tRNA methylation and formylation utilizing BS-Seq and fCAB-seq techniques. Their findings revealed that among all mitochondrial tRNAs, tRNA-Met exclusively carried the f5C modification. To ascertain f5C levels, fCAB-seq data was subtracted from BS-seq results, and the authors computed these levels by subtracting bisulfite-protected cytosines from ethylhydroxylamine-protected sites.
To delve further, the authors employed two distinct short hairpin RNAs (shRNAs) in four oral squamous cell carcinoma (OSCC) samples. Consequently, they observed a remarkable approximately 8-fold increase in unmodified cytosine at position C34 when the methyltransferase NSUN3 was depleted in these OSCC cell lines. Notably, minimal f5C modification was detected in mitochondrial tRNA-Met following NSUN3 depletion.
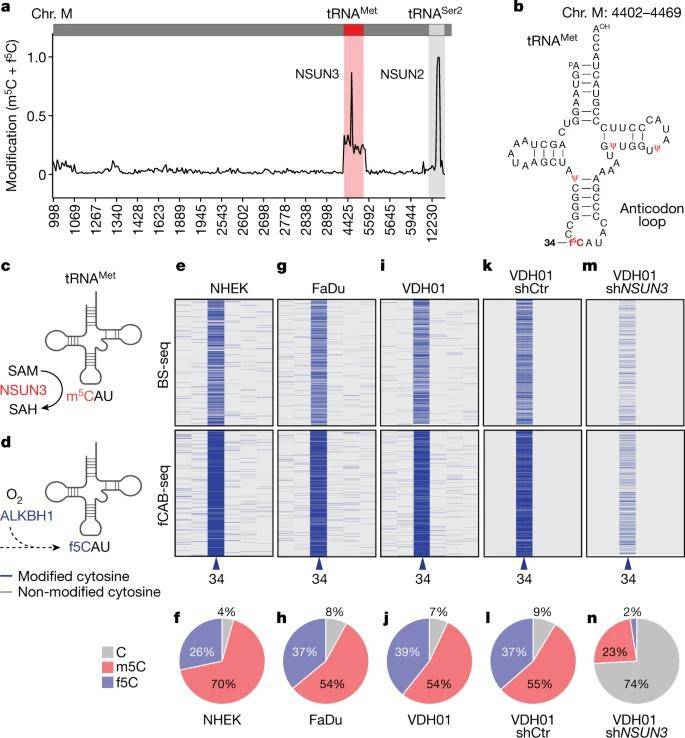 Detection of m5C and f5C in mitochondrial tRNAs. (Delaunay et al., 2022)
Detection of m5C and f5C in mitochondrial tRNAs. (Delaunay et al., 2022)
m5C Regulates Mitochondrial Function
The study's findings showcased a substantial decrease in de novo protein synthesis within NSUN3-depleted mitochondria, as evidenced by protein synthesis quantification experiments. Consequently, the hypomethylation of mitochondrial tRNAMet led to a down-regulation in protein levels of mitochondrial-encoded genes. To assess how mitochondrial metabolism adapts to this reduction in protein synthesis, the authors conducted mass spectrometry analysis to quantify tricarboxylic acid cycle (TCA) metabolites. Upon reducing NSUN3 expression, the study observed modest reductions in most TCA metabolites, mirroring the effects of NSUN3 depletion. Additionally, TCA metabolite levels were lower in cells lacking methylation. Consequently, maximal respiration decreased when tRNAMet was minimally modified, and NSUN3-deficient cancer cells exhibited an overall higher basal extracellular acidification rate (ECAR).
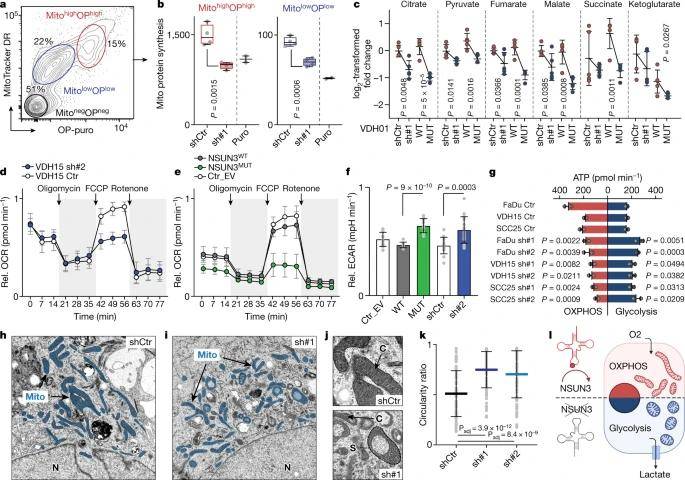 Mitochondrial m5C controls energy metabolism in tumor cells. (Delaunay et al., 2022)
Mitochondrial m5C controls energy metabolism in tumor cells. (Delaunay et al., 2022)
m5C Modification in Mitochondria Promotes Tumor Invasion
Immunostaining for mitochondrial markers, specifically MtCO1 and MtCO2, unveiled their robust expression within the basal CD44+ cell population. These CD44+ cells are notably characterized by poor differentiation and heightened proliferative potential, thus being deemed as metastatic precursor cells.
In an endeavor to elucidate the impact of inhibiting mitochondrial m5C formation on cell proliferation and invasion, the research team generated 3D cultures of oral squamous cell carcinoma (OSCC) cells. Notably, upon silencing NSUN3 and consequently inhibiting m5C modification, the dimensions of tumor-like cell clusters remained virtually unaltered, signifying that cell proliferation remained unaffected.
Of particular interest are CD44 and CD36, which serve as markers for metastatic-initiating cells in human oral cancer. Notably, CD36 is localized to the outer mitochondrial membrane. The subsequent investigation explored the co-expression of CD44 and CD36, shedding light on the association between initiating cells and mitochondrial function. Moreover, the research delved into lymph node metastasis within a subpopulation of CD44HCD36H cells. Notably, NSUN3 silencing led to a reduction in the abundance of CD44HCD36H cells.
Summary
The authors conducted a comprehensive assessment of m5C levels to predict metastasis in clinical patients. They employed histochemical staining on 78 human tumor samples to examine the correlation between NSUN3 protein expression and the pathological stage of the primary tumor.
Utilizing data from TCGA HNSCC patients, the authors investigated the differential expression of genes associated with NSUN3 levels. Notably, unsupervised clustering of patients resulted in the categorization of four distinct groups, revealing a progressive increase in both cancer stage and the presence of lymph node metastases at diagnosis with higher NSUN3 levels.
In summary, the characterization of NSUN3-driven genes proves to be a valuable predictor of lymph node metastasis and advanced pathological stages in HNSCC patients.
Furthermore, when mitochondrial translation is inhibited, it replicates the effects observed when NSUN3 protein function is silenced. This inhibition not only hinders cell invasion but also reduces the number of CD36-dependent metastasis-initiating tumor cells in both in vitro and in vivo settings. Consequently, targeting m5C formation within mitochondria emerges as a promising therapeutic avenue to halt the dissemination of tumor cells from the primary tumor site.
Reference:
- Delaunay, Sylvain, et al. "Mitochondrial RNA modifications shape metabolic plasticity in metastasis." Nature 607.7919 (2022): 593-603.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


