According to survey data from the World Health Organization, approximately 5 million children worldwide tragically lost their lives before reaching the age of 5 in 2021. Shockingly, 46% of these heart-wrenching deaths occurred within the first month of life, during what is known as the neonatal period. Traditionally, models for determining the causes of death in children have relied heavily on making inferences about the cause of death and analyzing vital registration data. However, these methods often attribute deaths to a single underlying disease, making it extremely challenging to unearth the true root cause of these heartbreaking losses.
In recent years, there has been a promising breakthrough in understanding the causes of death in children through a technique known as postmortem minimally invasive tissue sampling (MITS). By combining MITS with other valuable information, we can now delve deeper into characterizing the causes of neonatal death. This approach enables us to identify not only the immediate events leading to death but also the antecedents and underlying causes that ultimately result in these tragic outcomes.
Metagenomic Next-Generation Sequencing (mNGS)
Currently, conventional assays, such as traditional culture methods and nucleic acid amplification tests, have proven inadequate when it comes to detecting pathogens responsible for infectious deaths in children. Enter the next-generation metagenome sequencing (mNGS), which stands ready to tackle these challenges. Unlike its predecessors, mNGS does not rely on pre-selected targets and has the remarkable ability to identify viruses, bacteria, and fungi without any inherent bias. This advancement promises a more nuanced understanding of the role various pathogens play in the complex web of causes leading to death. Furthermore, mNGS empowers us to analyze antimicrobial drug resistance without the need for culture-based methods, allowing us to identify antimicrobial resistance genes present in samples.
In this research, the team assessed the effectiveness of mNGS in identifying pathogens associated with neonatal deaths. They did so by examining blood and lung tissue samples collected through MITS from postmortem neonates in Soweto, South Africa. Astoundingly, mNGS results aligned with those obtained through traditional culture methods and nucleic acid amplification testing (NAAT) in a staggering 90% of the cases, pinpointing the pathogens responsible for these tragic losses. Moreover, mNGS provided critical insights into the resistance profiles of these pathogens, unearthing many multi-drug resistant bacteria.
Metagenomic NGS Analysis Reveals Pathogens and Antimicrobial Resistance Genes in Neonatal Infections
The research team conducted a study involving 236 eligible neonates who unfortunately passed away. They carried out Minimally Invasive Tissue Sampling (MITS) on 153 of these neonates. The potential causes of death (CoDs) were diverse, with the majority (53%) attributed to complications arising from preterm birth, while 15% were linked to intrapartum events, and 13% resulted from congenital anomalies. Notably, 88 cases were associated with either confirmed or potential causes of infectious diseases.
Among the non-infectious disease-related deaths, 16 cases were included as controls. Among the patients who succumbed to infections, 31 had available residual lung tissue and blood samples for metagenomic next-generation sequencing (mNGS) analysis. In 20 of these cases, pathogens were already identifiable through routine ante-mortem or post-mortem cultures or nucleic acid amplification tests (NAAT), but for post-mortem samples only.
Analyzing the average number of mNGS sequencing reads or the percentage of non-host reads (reads not mapped to the human genome), the research team observed that control decedents had a lower percentage of non-host reads compared to cases with infectious CoDs. Remarkably, among the 20 cases with known pathogens, mNGS successfully identified the same pathogen in 18 cases, achieving an impressive 90% concordance. Furthermore, mNGS unveiled other microorganisms potentially responsible for neonatal deaths, such as Klebsiella aerogenes.
In 11 cases where deaths were attributed to infections that couldn't be preliminarily identified using conventional methods, mNGS played a pivotal role by identifying potential pathogens in 9 of these cases. The pathogens exclusively identified by mNGS included Klebsiella pneumoniae, Acinetobacter baumannii, and Streptococcus mucinis. Additionally, in 14 cases with positive antemortem blood cultures, mNGS consistently detected the same microorganisms in 86% of these cases.
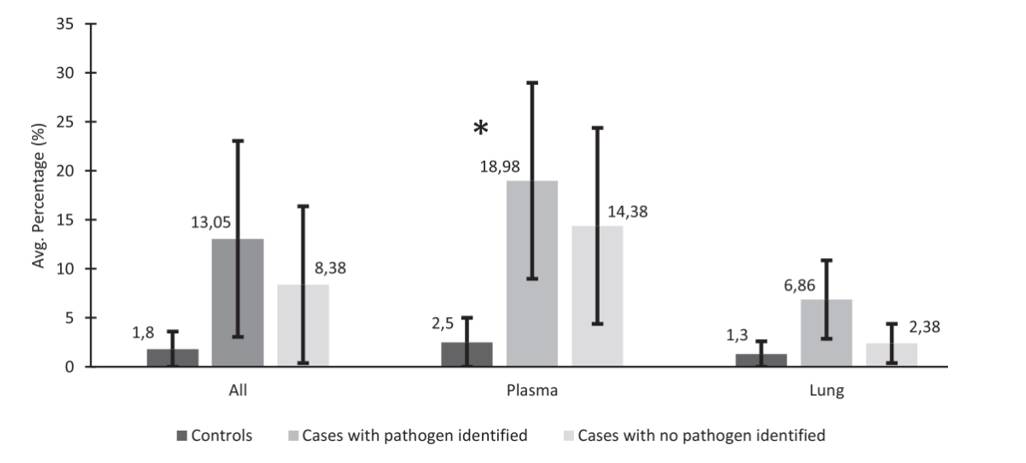 The metagenomic NGS metric for reads remaining after host and quality filtering. (Baillie et al., 2023)
The metagenomic NGS metric for reads remaining after host and quality filtering. (Baillie et al., 2023)
The research team meticulously analyzed the raw paired-end reads extracted from the mNGS RNA library to discern antibiotic resistance (AMR) genes. The findings revealed the presence of a substantial 32 AMR genes within the mNGS data, encompassing genes associated with resistance to β-lactams, aminoglycosides, sulfonamides, and others. Notably, the genes encoding β-lactamases exhibited remarkable diversity, with the identification of a total of 12 distinct mutations. Among these mutations, those found in the NDM-1 gene were the most prevalent.
Upon conducting a comprehensive assessment of AMR gene abundance, it became evident that cases where the pathogen was identified as the cause of death (CoD) displayed a higher number of AMR genes compared to infectious deaths in which the pathogen remained unidentified.
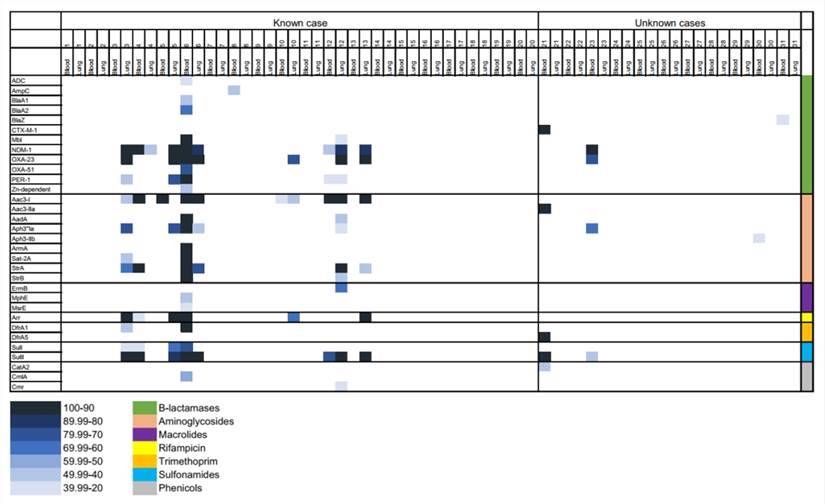 Antimicrobial resistance (AMR) genes in samples from neonates that died of known and unknown pathogens using RNA libraries. (Baillie et al., 2023)
Antimicrobial resistance (AMR) genes in samples from neonates that died of known and unknown pathogens using RNA libraries. (Baillie et al., 2023)
The research team meticulously prepared DNA libraries for a total of 18 cases, categorizing them into two distinct groups: one consisting of 12 cases with identified pathogens and the other comprising 6 cases where the pathogens remained unidentified. Within the subset of cases with known pathogens, the DNA libraries proved their mettle by successfully identifying potential causative agents. In six instances, the infection-associated microorganism was unequivocally pinpointed, with all cases revealing the presence of Acinetobacter baumannii, either through culture or nucleic acid amplification testing (NAAT).
Subsequently, the team delved deeper into the six instances of A. baumannii positive samples mentioned earlier. Through their diligent efforts, they managed to generate comprehensive genome sequences with remarkable completeness, ranging from 92% to 99%. Strikingly, all of these genomes fell within the closely related ST1 type, underlining the genetic homogeneity of these strains.
Notably, amidst the cases where no pathogen had initially been detected, the DNA library proved its worth by unveiling a single instance of Klebsiella pneumoniae.
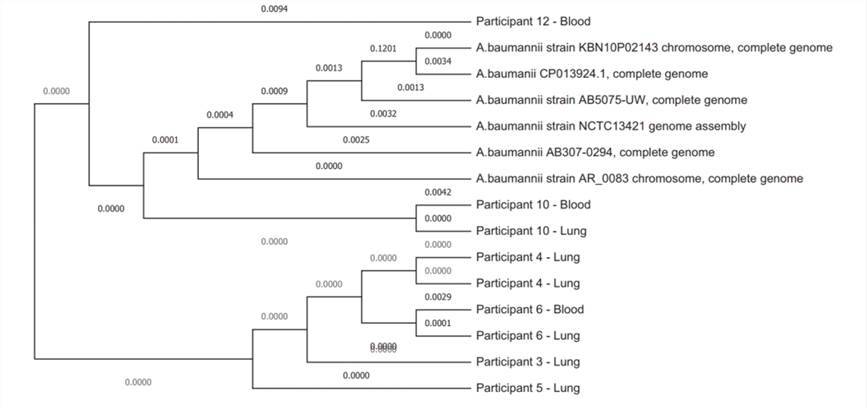 Whole genome phylogeny of the genomes of the 9 Acinetobacter baumannii strains collected from 6 neonatal deaths evaluated in the DNA libraries for this study together with 6 reference strains. (Baillie et al., 2023)
Whole genome phylogeny of the genomes of the 9 Acinetobacter baumannii strains collected from 6 neonatal deaths evaluated in the DNA libraries for this study together with 6 reference strains. (Baillie et al., 2023)
Subsequently, the research team conducted a thorough analysis of raw paired-end DNA library reads obtained from the identified microbial samples, focusing on the detection of antimicrobial resistance (AMR) markers. In this comprehensive analysis, they uncovered a total of 29 distinct AMR genes exhibiting resistance to β-lactams, aminoglycosides, and sulfonamides. Notably, among these genes, those encoding β-lactamases and aminoglycosides displayed remarkable diversity, with each category comprising nine distinct genes.
 Antimicrobial resistance (AMR) genes in samples from neonates that died of infectious diseases using DNA libraries. (Baillie et al., 2023)
Antimicrobial resistance (AMR) genes in samples from neonates that died of infectious diseases using DNA libraries. (Baillie et al., 2023)
The findings from this study strongly indicate that employing clinical metagenomic Next-Generation Sequencing (NGS) with postmortem minimally invasive tissue sampling (MITS) could prove invaluable for pinpointing pathogen-specific causes of neonatal infection-related deaths. Furthermore, this pioneering diagnostic approach holds the potential to significantly inform treatment strategies within neonatal intensive care units, facilitate the early detection of emerging infections and disease profiles, and play a vital role in both identifying and helping control outbreaks of hospital-acquired infections. Moreover, in resource-constrained countries or regions, the application of mNGS offers a promising avenue for assessing disease-specific pathogen prevalence and identifying the prevailing antimicrobial drug resistance gene profiles, thereby guiding the targeted development of pharmaceutical interventions.
Reference:
-
Baillie, Vicky L., et al. "Metagenomic sequencing of post-mortem tissue samples for the identification of pathogens associated with neonatal deaths." Nature Communications 14.1 (2023): 5373.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


