Research Background
The composition of the human microbiome is strongly influenced by various lifestyles. However, current microbiome research predominantly centers on Western industrialized populations, which exhibit notably low microbiome diversity. To unravel the intricate microbiome alterations driven by industrialized lifestyles, an extensive investigation involving high-depth metagenomic sequencing is imperative. They undertook a pioneering study to delve into microbial function, growth kinetics, and dispersal patterns in a unique population of primitive humans, the Hadza. The research involved sequencing a remarkable 351 Hadza fecal samples, resulting in an impressive 9.39 terabytes of metagenomic data and yielding a total of 83,044 metagenome-assembled genomes (MAGs), along with associated bioinformatic findings.
Experimental Design
The Hadza, one of the last surviving populations in Africa preserving the foraging traditions of our human ancestors, served as the focal point of our study. The research efforts included collecting 351 fecal samples from 167 individuals, processing and sequencing 388 DNA samples, and obtaining 52 purified bacterial isolates. The microbiome composition of all samples was meticulously assessed, employing the newly reconstructed genomes from our study. The metagenomic data was meticulously aligned with a series of customized databases that contained the complete genome sequences of species-level representatives across bacterial/archaeal (n=5,755), phage (n=68,461), and eukaryotic (n=12) domains.
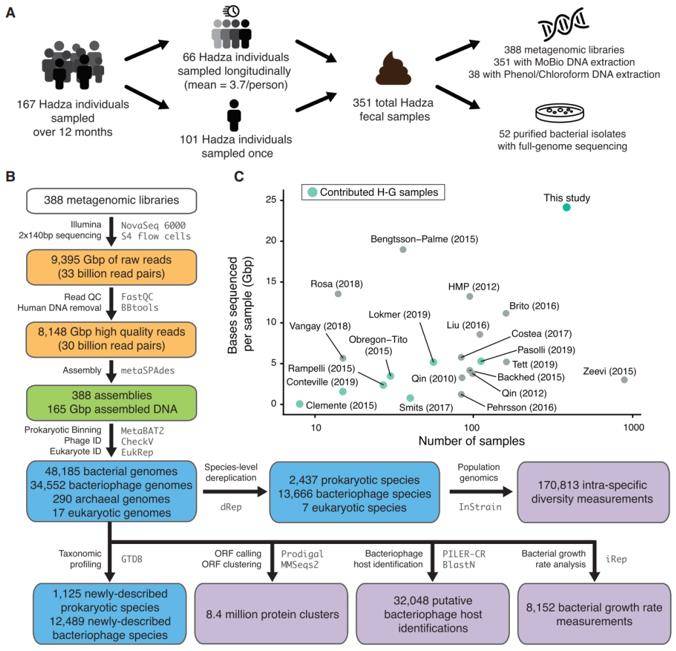 A vast resource of the Hadza gut microbiome data. (Carter et al., 2023)
A vast resource of the Hadza gut microbiome data. (Carter et al., 2023)
Hadza Gut Microbial Diversity
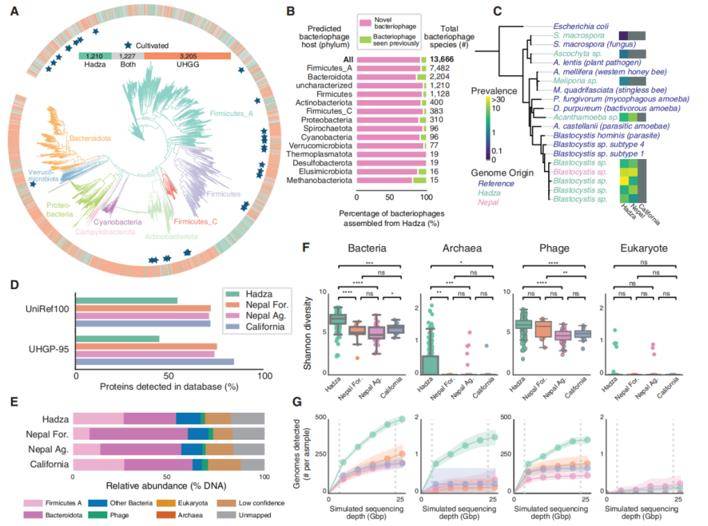
In this investigation, the gut microbial diversity of the Hadza population was explored, expanding the dataset with information from the Unified Human Gastrointestinal Genome (UHGG) and the Metagenomic Global Catalog (MGV). Notably, the Hadza microbiome contained 20.2% of previously unlisted microorganisms, underscoring the substantial depth of microbial diversity within this population. In contrast, 79.9% of the microorganisms in the Hadza microbiome were recognized species.
Moreover, this study exposed the presence of previously unknown eukaryotic microorganisms within the Hadza gut microbiome. A comprehensive annotation identified an impressive 8.4 million protein families, with 59.7% of them absent from the Unified Human Gastrointestinal Protein (UHGP) database.
To gain further insights, a comparative analysis was performed, contrasting the Hadza microbiome with fecal samples from Nepalese and Californian populations, utilizing a deep metagenomic sequencing approach. The assessment of microbiome data across bacterial/archaeal (n=5,755), phage (n=68,461), and eukaryotic (n=12) domains was conducted using three customized metagenome-assembled genome (MAG) databases.
 The Hadza gut microbiota contains substantial multi-domain novelty. (Carter et al., 2023)
The Hadza gut microbiota contains substantial multi-domain novelty. (Carter et al., 2023)
The findings indicated a significantly higher diversity of bacteria, archaea, and phages within the Hadza population compared to other populations studied. In summary, the Hadza samples exhibited greater richness in bacteria, archaea, phages, and unclassified species at various sequencing depths when compared to their counterparts in other populations, highlighting the exceptional microbial diversity within the Hadza population.
 VANISH and BloSSUM taxa have distinct global prevalence, phylogeny, and functional capacity. (Carter et al., 2023)
VANISH and BloSSUM taxa have distinct global prevalence, phylogeny, and functional capacity. (Carter et al., 2023)
Disappearing Populations in the Face of Industrialization Detected
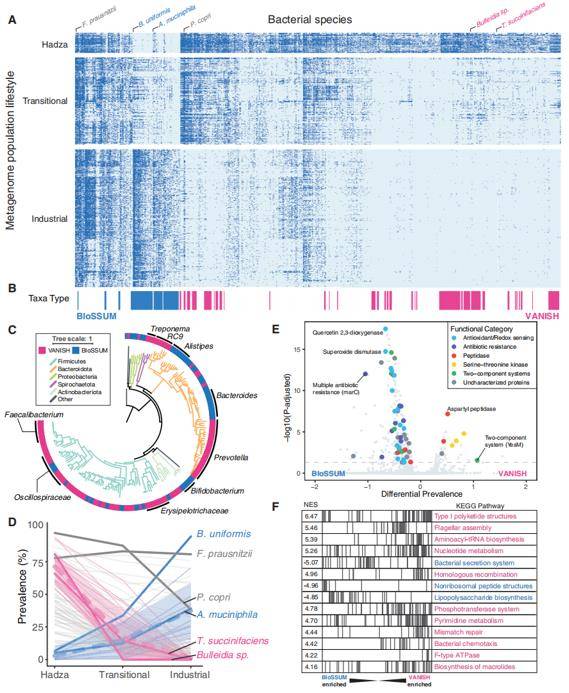
This study incorporated a dataset of 1,800 data points, comprising 950 samples from industrialized populations, 583 samples from transitional populations, 135 samples from the Hadza, and 132 samples from other hunter-gatherer communities, sourced from 18 published studies. These data were categorized into two taxa, VANISH (indicating microorganisms that vanished during industrialization, totaling 124 species) and BloSSUM (signifying microorganisms selected in the industrialization era, encompassing 63 species). Further analyses uncovered a compelling correlation between the phylogeny and the industrialization process. Intriguingly, the BloSSUM taxa seemed to exhibit a competitive edge over the VANISH taxa within the transitional human gut environment.
Additionally, individual KEGG-based analyses revealed 37 and 268 genes shared by the VANISH and BloSSUM taxa, respectively. The VANISH taxa were notably linked to peptidase and spore-producing genes, while the BloSSUM taxa showed a distinct association with antioxidant and redox-related genes. The study systematically grouped these genes into pathways, facilitating a comprehensive set enrichment analysis. The observed variations in gene functions and enriched pathways between the two taxa underscored the functional non-redundancy of the BloSSUM taxa compared to the VANISH taxa.
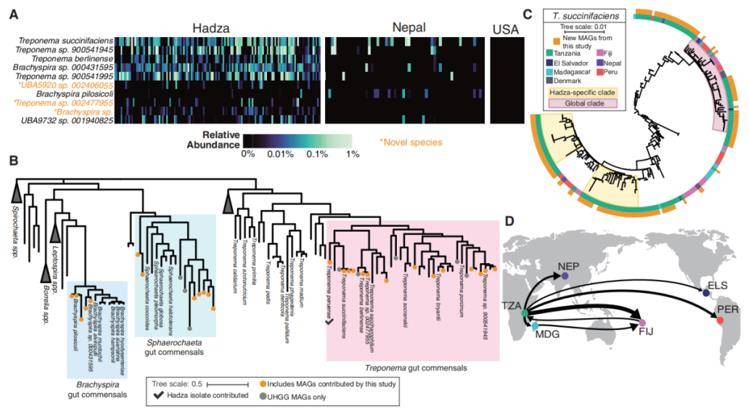 Spirochaetota that are highly abundant in the Hadza are absent in industrial samples. (Carter et al., 2023)
Spirochaetota that are highly abundant in the Hadza are absent in industrial samples. (Carter et al., 2023)
Evolution, Replication, and Dispersal of Gut Microbes in the Hadza Population
The adoption of deeper sequencing and novel metagenomic approaches reaffirmed the seasonal cycling of gut microbe composition and carbohydrate-active enzymes in the Hadza population. Additionally, the study, for the first time, quantified the seasonal cycling in the in situ replication rates of gut microbes in the Hadza population.
To identify genes experiencing distinct selective pressures within the Hadza microbiota, we analyzed whole gene pN/pS ratios. This analysis revealed instances of positive or diverse selection, likely influenced by dynamic changes within the gut environment, such as seasonal shifts in diet, host immune system responses, and strategies employed by microbes to evade viral intrusion.
Furthermore, an ANI analysis was conducted to explore the relatedness of strains among familial and non-familial individuals. The results indicated that family members shared a higher number of homologous strains than non-familial individuals, and they also shared more closely related strains than unrelated individuals. This underscores the significance of familial connections and genetic relatedness in shaping the Hadza gut microbial landscape.
Reference:
- Carter, Matthew M., et al. "Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes." Cell (2023).
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


