The orchestration of gene expression in plants involves intricate interactions between chromatin structure and transcriptional activity. The advent of high-throughput sequencing techniques, particularly ATAC-seq and RNA-seq, has revolutionized the study of gene regulation. ATAC-seq enables the mapping of open chromatin regions, providing insights into the accessibility of DNA and the localization of regulatory elements. In contrast, RNA-seq quantifies gene expression levels, shedding light on the dynamic transcriptional landscape. Integrating these two datasets empowers researchers to unravel the interplay between chromatin architecture and gene expression.
Integrating ATAC-seq and RNA-seq
ATAC-seq captures open chromatin regions by utilizing a transposase enzyme to fragment accessible DNA. This technique identifies areas where transcription factors and other regulatory proteins bind, influencing gene expression. By analyzing ATAC-seq data, researchers can pinpoint enhancers, promoters, and other cis-regulatory elements that govern gene activation or repression.
Integrating ATAC-seq and RNA-seq data provides a holistic perspective on gene regulation. It allows researchers to connect chromatin accessibility with gene expression dynamics, uncovering causal relationships between regulatory elements and their target genes. This integration aids in identifying potential transcription factors that drive specific expression patterns by modulating chromatin structure. Furthermore, it enables the identification of co-regulated gene modules that play essential roles in cellular processes.
Analytical Strategies
The integration of ATAC-seq and RNA-seq data involves a multi-step analysis. After individual data processing, normalization, and quality control steps, the datasets are integrated through statistical correlations or machine learning approaches. Correlation analysis identifies genes with concordant changes in chromatin accessibility and expression. Differential expression analysis on both datasets can uncover genes undergoing coordinated changes, shedding light on their functional relevance.
Case Studies
Case Study 1: Wheat Regeneration
Genetic transformation is indispensable for investigating gene function and enhancing crop traits. However, this technique is less efficient in wheat. To address this limitation, a holistic understanding of the transcriptional and chromatin dynamics during wheat regeneration is necessary. The integration of ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) and RNA-seq (RNA sequencing) techniques offers a powerful means to unravel the gene regulatory network orchestrating regeneration.
RNA-seq, ATAC-seq and CUT&Tag techniques.
The integrated multi-omic analysis revealed that the auxin-mediated sequential expression of genes governing cell fate transition is closely tied to changes in chromatin accessibility and histone modifications. The constructed TRN driving wheat regeneration was dominated by 446 essential transcription factors. Comparative analysis highlighted divergent DNA binding patterns of DOF TFs between wheat and Arabidopsis. Experimental validation demonstrated that TaDOF5.6 and TaDOF3.4 could enhance transformation efficiency in distinct wheat varieties, underscoring their potential role in improving genetic transformation efficacy.
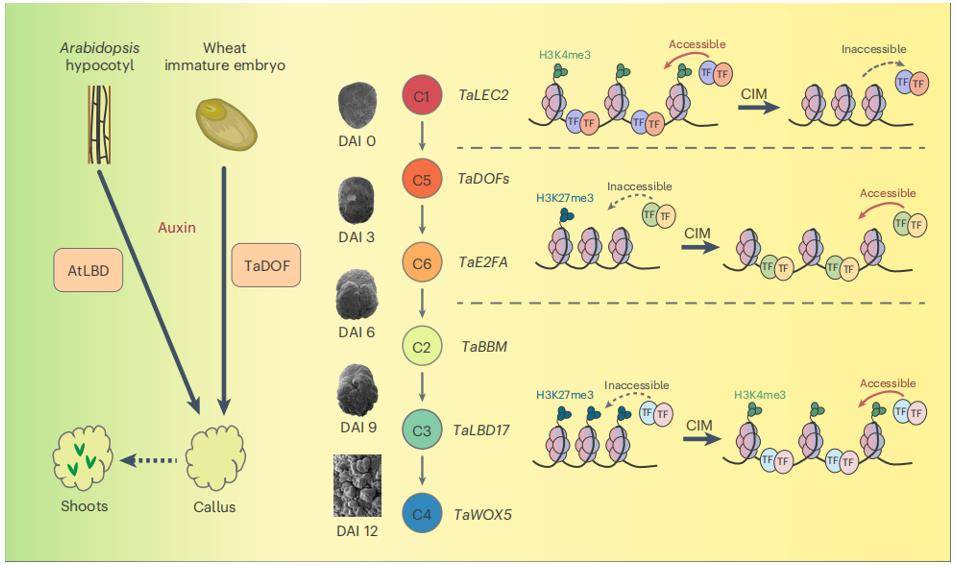 Wheat regeneration and transformation. (Liu et al., 2023)
Wheat regeneration and transformation. (Liu et al., 2023)
Case Study 2: Gene Expression Patterns in Plant Polyploids
The adaptability and plasticity of allohexaploid bread wheat (Triticum aestivum. L., BBAADD) are thought to be influenced by expression divergence due to genetic variation and interactions among its subgenomes. However, the underlying molecular mechanisms of this expression divergence remain unclear. Squamosa promoter-binding protein-like (SPL) transcription factors play pivotal roles in various biological processes. To elucidate the regulatory network governing gene expression in wheat and the role of SPLs in this process, this study integrated various omics techniques, including DAP-seq for 40 SPLs, ATAC-seq, and RNA-seq.
DAP-seq、ATAC-seq and RNA-seq
Diverse SPL Targeting: The study revealed that a set of low-affinity SBRs were targeted by multiple SPLs, leading to differential sequence preferences around the core GTAC motif.
Conservation of SBRs: Despite their low affinity, the identified SBRs were evolutionarily conserved and showed enrichment for GWAS signals related to important agricultural traits.
CRE Variations and Subgenome Divergence: Analysis of the SBRs within the cis-regulatory regions (CREs) of syntenic genes demonstrated high diversification among the subgenomes. Only a small fraction of SBRs (less than 8%) coexisted in triad genes, emphasizing the critical role of CRE variations in subgenome differentiation.
Functional Validation: Knockout experiments of TaSPL7A/B/D and TaSPL15A/B/D subfamilies provided experimental evidence that both high- and low-affinity SBRs play essential roles in regulating gene expression for controlling tiller number and spike sizes.
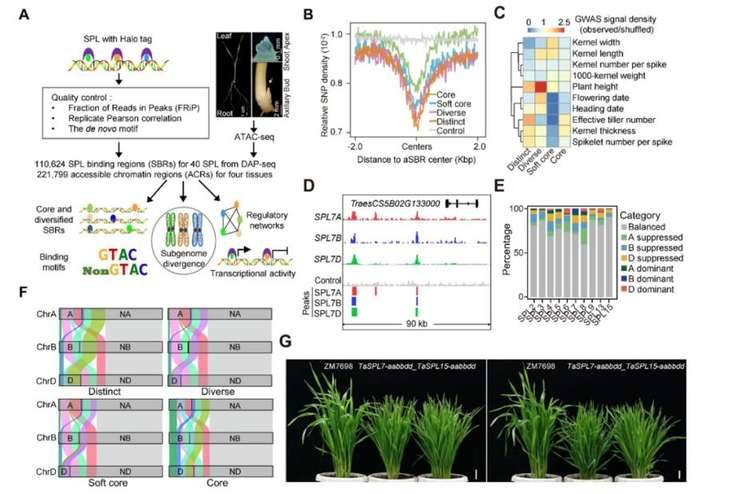 Low-affinity SPL binding sites contribute to subgenome expression divergence in allohexaploid wheat. (Pei et al., 2023)
Low-affinity SPL binding sites contribute to subgenome expression divergence in allohexaploid wheat. (Pei et al., 2023)
Case Study 3: Plant Heat Stress Responses
Background:
The intricate regulation of gene expression in plants involves complex interactions between transcription factors (TFs), chromatin structure, and the three-dimensional organization of the genome within the nucleus. This organization provides plants with the ability to rapidly respond to environmental stresses by altering gene expression patterns. However, the mechanisms underlying the dynamic changes in chromatin conformation and their relationship to gene expression during stress responses remain poorly understood. This case study focuses on investigating the genome-wide chromatin changes associated with transcriptional reprogramming in response to heat stress in tomato plants.
Methods
To elucidate the dynamics of chromatin reorganization and gene expression under heat stress conditions, the researchers utilized a combination of two powerful techniques: Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) and RNA sequencing (RNA-seq).
Results
The data showed that heat stress induced rapid changes in the chromatin architecture of tomato plants. These changes involved the transient formation of promoter-enhancer contacts, suggesting a role for 3D chromatin conformation in mediating stress-responsive gene expression. The differentially expressed genes identified through RNA-seq indicated that the observed chromatin changes were associated with the expression of heat-stress responsive genes. This suggested a direct link between altered chromatin architecture and the activation of specific genes necessary for heat stress adaptation.
The researchers further explored the molecular mechanisms driving chromatin reorganization during heat stress. They found that the transcription factor HSFA1a, known to be essential for heat stress tolerance in tomato, played a crucial role in orchestrating chromatin spatial reorganization. This indicated that transcription factors are key players in controlling dynamic transcriptional responses through 3D reconfiguration of promoter-enhancer contacts.
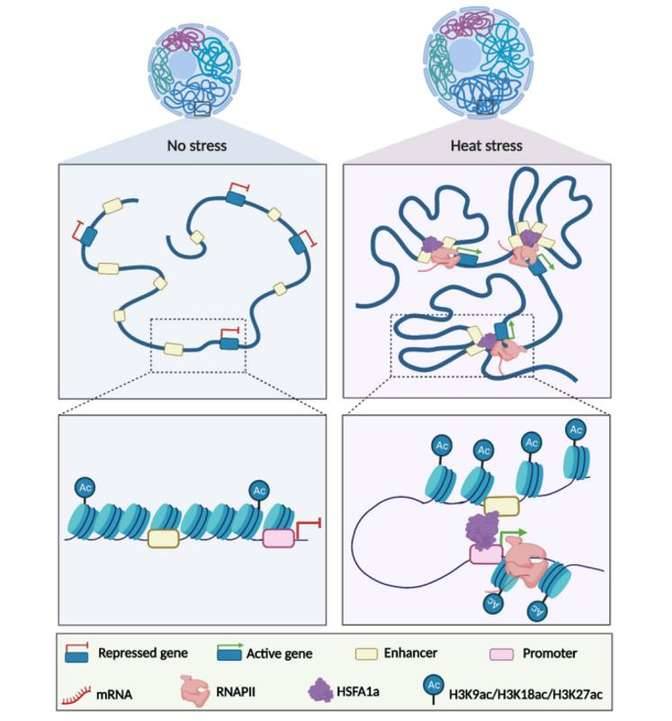 HSFA1a modulates plant heat stress responses and alters the 3D chromatin organization of enhancer-promoter interactions. (Huang et al., 2023)
HSFA1a modulates plant heat stress responses and alters the 3D chromatin organization of enhancer-promoter interactions. (Huang et al., 2023)
References:
-
Liu, Xuemei, et al. "Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation." Nature Plants (2023): 1-18.
- Pei, Hongcui, et al. "Low-affinity SPL binding sites contribute to subgenome expression divergence in allohexaploid wheat." Science China Life Sciences 66.4 (2023): 819-834.
- Huang, Ying, et al. "HSFA1a modulates plant heat stress responses and alters the 3D chromatin organization of enhancer-promoter interactions." Nature Communications 14.1 (2023): 469.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


