
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
CRISPR/Cas9 nucleases have emerged as a revolutionary tool in genetic engineering, offering precise modifications across a wide range of organisms, including major crops. This review examines the mechanisms underlying CRISPR/Cas9 off-target effects, the initial studies highlighting these issues, and the ongoing efforts to mitigate them. Various detection methods, such as whole-genome sequencing (WGS), Breaks Labeling, Enrichment on Streptavidin, and Sequencing (BLESS), Genome-Wide, Linear amplification-mediated high-throughput genome-wide translocation sequencing (LAM-HTGTS), Digenome-seq, and SITE-Seq, are explored for their effectiveness in identifying unintended genomic alterations. Additionally, computational tools for single guide RNA (sgRNA) target finding and off-target prediction are discussed. Despite the challenges posed by off-target effects, significant advancements in understanding and reducing these effects have been made, enhancing the specificity and safety of CRISPR/Cas9 applications. This review emphasizes the importance of continued research and innovation to ensure the reliable use of CRISPR/Cas9 in both research and clinical settings.
CRISPR/Cas9 nucleases have markedly advanced genetic engineering by facilitating precise genetic modifications across a wide range of organisms. This technology leverages the adaptive immune system of prokaryotes to selectively target and modify specific DNA sequences, thus serving as a highly versatile genome editing tool. However, a notable limitation of this system is the potential for off-target mutations—unintended genomic alterations that occur when the CRISPR/Cas9 complex erroneously binds to and cleaves non-target DNA sequences. These off-target effects compromise the accuracy and reliability of CRISPR/Cas9-mediated gene editing, as highlighted by Cradick et al. (2013).
While some studies suggest CRISPR is more prone to unintended cleavage events compared to zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), its versatility and ease of use have quickly made CRISPR the preferred genome editing tool (Gao et al. 2019; Huang et al. 2018; Karimian et al. 2019). This review aims to provide an overview of the CRISPR/Cas9 system, discuss the types of off-target effects, and explore various methods and tools developed for detecting and predicting these unintended consequences. Addressing these challenges is crucial for enhancing the specificity and reliability of CRISPR/Cas9, thereby facilitating its safe and effective application across various fields, including agricultural biotechnology and human gene therapy.
The CRISPR/Cas9 system, a groundbreaking tool in genome editing, has its origins in the study of bacterial adaptive immunity. Tandemly repeated sequences were first discovered in 1987 during research on the Escherichia coli alkaline phosphatase (iap) gene (Ishino et al. 1987). These sequences were named clustered regularly interspaced short palindromic repeats (CRISPR) in 2002, with the adjacent genes designated as Cas (CRISPR-associated) (Jansen et al. 2002). By 2007, it was confirmed that CRISPR and Cas proteins play a crucial role in the adaptive immune mechanisms of bacteria (Barrangou et al. 2007). The CRISPR/Cas9 system achieved its first high-efficiency editing of endogenous genes in human and mouse cells in 2013 (Cong et al. 2013).
Many bacteria and most archaea employ the CRISPR/Cas system, present in their genomes or plasmids, to defend against invading viruses or bacteriophages. When a virus or bacteriophage injects genetic material into a bacterium, Cas proteins integrate a small segment of the foreign DNA into the CRISPR sequence at the 5' end, creating an "immunological memory" of the invader. Upon subsequent encounters, the bacterium transcribes this CRISPR sequence into single guide RNA (sgRNA), which complexes with Cas proteins to specifically cleave the foreign DNA. This targeted cleavage silences the invader, thereby granting the bacterium immunity against the virus or bacteriophage (Horvath & Barrangou 2010).
Off-target effects in the CRISPR/Cas9 system have been classified into three primary types. The first type involves regions at other protospacer adjacent motifs (PAMs) (5'-NGG-3') that have single or multiple base substitutions (Fu et al. 2013). The second type includes regions at other PAMs (5'-NGG-3') containing insertions and/or deletions (indels) compared to the target DNA or RNA spacer (Fig. 1). These indels can form small bulges, allowing them to anneal correctly and facilitate Cas9 activity, sometimes resulting in higher off-target activities than on-target ones (Lin et al. 2014). The third type involves sequences with different PAM sites (5'-NAG-3') (Tsai et al. 2015).
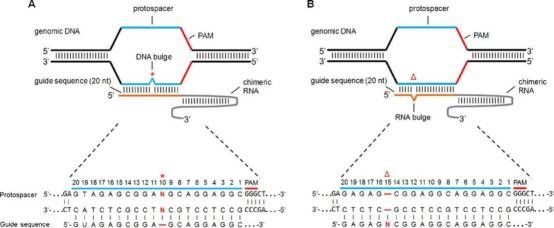 Fig.1 Schematic of CRISPR/Cas9 off-target sites with (A) 1-bp insertion (DNA bulge) or (B) 1-bp deletion (RNA bulge) (Lin et al. 2014)
Fig.1 Schematic of CRISPR/Cas9 off-target sites with (A) 1-bp insertion (DNA bulge) or (B) 1-bp deletion (RNA bulge) (Lin et al. 2014)
WGS provides a comprehensive and unbiased approach to identify mutations across the entire genome, making it valuable for detecting off-target effects from CRISPR/Cas9 systems (Boyle et al. 2017). The process involves creating a library of mutant targets on a parallel array and assessing the dCas9–sgRNA complex binding in real-time using fluorescent labeling (Fig. 2). This library, based on a 20-bp phage λ-target sequence with various double substitutions, is sequenced using Illumina technology to map sequence clusters. Subsequently, Cy3-labeled RNP complexes are introduced into a modified GAIIx instrument, and their binding is monitored through fluorescence imaging. This setup allows for the empirical measurement of dCas9 binding dynamics at different target sites.
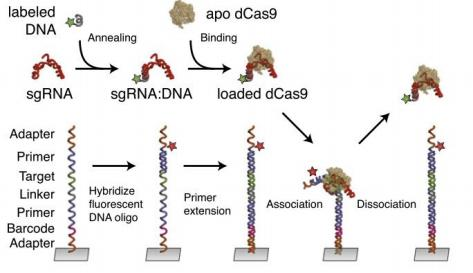 Fig.2 Experimental procedure for high-throughput biochemical profiling (Boyle et al. 2017)
Fig.2 Experimental procedure for high-throughput biochemical profiling (Boyle et al. 2017)
Despite its strengths, WGS has limitations in sensitivity, often detecting only higher-frequency off-target mutations in bulk cell populations (Veres et al. 2014). It also faces challenges in distinguishing single nucleotide variants (SNVs) from sequencing errors or natural variations, underscoring the need for a thorough genetic background investigation before evaluating off-target effects (Ishida et al. 2015). Nevertheless, WGS remains a potent tool for detecting all types of off-target base editing when applied to genomic DNA from multiple independent cells or whole organisms (Martin et al. 2016).
BLESS is an effective method for detecting double-strand breaks (DSBs) across the genome in mouse and human cells, minimizing false positives by avoiding artificial DSB formation during DNA extraction (Crosetto et al. 2013). It has been used to identify DSBs induced by the Sce endonuclease and study complex DSB landscapes and telomere ends. Off-target mutations from nuclease-induced DSBs, repaired by error-prone non-homologous end joining (NHEJ), can be tracked using BLESS. This method involves ligating break ends to biotinylated linkers, capturing fragments with streptavidin, adding a second barcoded linker, and identifying products through PCR and sequencing (Fig. 3). By labeling the DSB itself, BLESS offers an unbiased, genome-wide identification of DSBs. Studies have shown low off-target activity in mice and human cells, although BLESS only detects breaks present at the time of labeling but not those already repaired (Ran et al. 2015; Slaymaker et al. 2016; Tsai & Joung 2016).
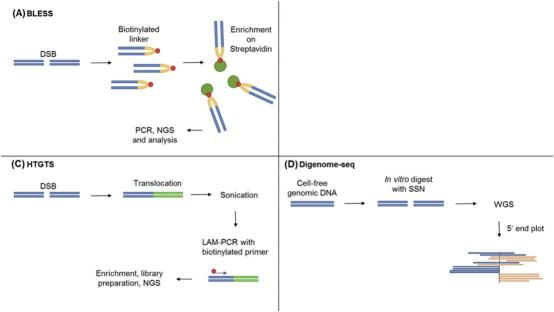 Fig.3 Methods for the detection of off-target mutations caused by SSNs. (A) BLESS: Captures DSBs with a biotinylated linker, enriches on streptavidin, and identifies by PCR and NGS. (C) HTGTS: Involves translocation with a bait DSB, sonicates DNA, amplifies the bait sequence, enriches, and analyzes byNGS. (D) Digenome-seq: Digests cell-free genomic DNA in vitro with SSNs, performs WGS, and identifies cleaved sites by 5' end plotting. (Zischewski et al. 2017)
Fig.3 Methods for the detection of off-target mutations caused by SSNs. (A) BLESS: Captures DSBs with a biotinylated linker, enriches on streptavidin, and identifies by PCR and NGS. (C) HTGTS: Involves translocation with a bait DSB, sonicates DNA, amplifies the bait sequence, enriches, and analyzes byNGS. (D) Digenome-seq: Digests cell-free genomic DNA in vitro with SSNs, performs WGS, and identifies cleaved sites by 5' end plotting. (Zischewski et al. 2017)
LAM-HTGTS tracks genomic translocations resulting from end-joining between DSBs (Frock et al. 2015). It detects DSBs induced by site-specific nucleases (SSNs) like TALENs and Cas9 through translocation to a known bait DSB (Hu et al. 2016). The nuclease cleaves the bait sequence, leading to chromosomal translocations when the break fuses with another DSB. Linear amplification PCR with a bait-specific primer amplifies the translocated sequence from bulk genomic DNA. Barcodes and adapters are added for NGS analysis (Fig. 3). Non-translocated DSBs can be selectively digested to prevent their sequencing. Although standard LAM-HTGTS does not identify small indels or single nucleotide polymorphisms (SNPs), it can be modified accordingly. LAM-HTGTS screens for large genomic rearrangements from nuclease-induced DSBs but requires simultaneous bait and other DSBs.
Digenome-seq leverages Cas9 as an in vitro nuclease to create an unbiased profile of off-target effects from selected gRNAs by digesting cell-free genomic DNA and sequencing the resulting fragments (Fig. 3). This method identifies Cas9-induced breaks by aligning sequence reads with identical ends, enabling the detection of off-target sites with mutation frequencies below 0.1% (Kim et al. 2015). Digenome-seq is cost-effective, capable of analyzing up to 10 gRNAs per run, and avoids issues related to random DSBs in cells and DNA repair processing (Kim et al. 2016). However, differences in Cas9 activity between in vitro and in vivo conditions may lead to false positives or negatives (Fu et al. 2016). Despite these challenges, Digenome-seq remains a sensitive and robust method for genome-wide profiling of off-target effects, capturing potential off-target sites with RNA/DNA bulges and detecting induced indels at very low frequencies.
Several additional methods are employed to detect off-target effects in gene editing. SITE-Seq (Selective Integration of Targeted Endonuclease Sequencing) uses a biochemical strategy to identify Cas9 cleavage sites in purified genomic DNA, enabling specificity profiling with minimal read depth (Cameron et al. 2017). DISCOVER-Seq (Discovery of in vivo CRISPR Edits by sequencing) is a sensitive assay for identifying off-target sites in cellular models, even in vivo, during therapeutic gene editing (Wienert et al. 2019). GOTI (Genome-wide Off-Target Analysis by Two-cell embryo Injection) evaluates off-target effects induced by various gene-editing tools in early-stage mouse embryos and derived cell populations, without interference from SNPs (Zuo et al. 2019). EndoV-seq (Endonuclease V Sequencing) uses Endonuclease V to assess the genome-wide specificity of adenine base editors (ABE) by digesting deaminated DNA before whole-genome sequencing (Liang et al. 2019). Lastly, VIVO (Verification of in vivo Off-targets) robustly detects genome-wide off-target effects in vivo, demonstrating efficient editing without detectable off-target mutations when using well-designed gRNAs (Akcakaya et al. 2018). These methods provide diverse and comprehensive approaches to understanding and mitigating off-target effects in gene editing.
Take the Next Step: Explore Related Services
A variety of experimental systems are widely used to investigate and predict off-target effects of sgRNAs in gene editing. Many algorithms have been developed to predict off-target effects and enhance the specificity of Cas9 by minimizing off-target activity. Sequencing-based approaches are employed to validate Cas9 off-targets experimentally, raising questions about the mechanisms of Cas9 binding and cleavage of off-targets. Various tools have been developed based on mismatch information, including PEM-seq (Primer-Extension-Mediated Sequencing) for detecting off-target effects and determining CRISPR/Cas9 specificity (Yin et al. 2019), CRISPR-PLANT v2 for designing gRNAs in plants (Minkenberg et al. 2019), CCTop (CRISPR/Cas9 Target Online Predictor) for identifying and ranking sgRNA target sequences (Stemmer et al. 2015), and CROP-IT (CRISPR/Cas9 Off-Target Prediction and Identification Tool) for improved site predictions of Cas9 binding and cleavage (Singh et al. 2015). Other notable tools include CHOPCHOP for selecting target sequences (Labun et al. 2016, Montague et al. 2014), the CFD (Cutting Frequency Determination) score for scoring mismatch positions (Doench et al. 2016), CT-Finder (CRISPR Target Finder) for precise target prediction (Zhu et al. 2016), CRISPOR for gRNA selection (Haeussler et al. 2016), CRISPR-GE for comprehensive solutions in plants (Xie et al. 2017), an ensemble learning method for efficient off-target site prediction (Peng et al. 2018), and Cas-OFFinder for versatile off-target mutation detection (Bae et al. 2014). These tools provide robust solutions for designing and evaluating sgRNAs, contributing to more precise and effective gene editing.
CRISPR/Cas9 technology has significantly revolutionized genetic engineering by providing unparalleled precision and flexibility in genome editing. Nonetheless, the presence of off-target effects constitutes a substantial challenge to its widespread adoption. The advancement of various detection methodologies and predictive algorithms has markedly improved the understanding and mitigation of these unintended alterations. Ongoing research and technological innovations are imperative for enhancing the specificity and dependability of CRISPR/Cas9, thereby ensuring its safe and effective application across diverse domains, including agricultural biotechnology and therapeutic gene editing. These collaborative efforts are anticipated to result in more precise and efficient utilization of CRISPR/Cas9, ultimately unlocking its full potential in scientific research and medical interventions.
References
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.


