
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
The CRISPR-Cas9 system, a pioneering RNA-guided genome-editing tool, has seen widespread application in biomedical research. Despite its utility, concerns regarding the specificity of Cas9 nuclease across the genome persist. To address these concerns, a study published in Nature Methods in 2014 introduced a novel methodology for off-target site identification: Digenome-seq (Digenome Sequencing), an approach for genome-wide profiling of CRISPR-Cas9 off-target effects in human cells.
Digenome-seq is predicated on in vitro digestion of the genome using Cas9, followed by high-throughput sequencing. The generated sequences, which exhibit a 5' end identical to the 5' end of Cas9 cleavage sites, can be identified through computational methods. This technique enables the detection of off-target sites with an insertion/deletion (indel) frequency as low as 0.1%, approaching the detection limits of targeted deep sequencing.
The study demonstrated that Cas9 nuclease possesses high specificity, inducing off-target mutations at only a few locations across the entire genome rather than thousands of sites. Furthermore, it was observed that Cas9 off-target effects can be mitigated by replacing sgRNAs with those exhibiting improved specificity. Digenome-seq is thus affirmed as a reliable, sensitive, unbiased, and cost-effective method for analyzing the genome-wide off-target effects of programmable nucleases, including Cas9.
Programmable nucleases, such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided engineered nucleases (RGENs) derived from the type II CRISPR-Cas system, serve as invaluable tools for genome editing in cultured cells and whole organisms. Nevertheless, these nucleases are also associated with the induction of off-target mutations. For example, the RGEN complex consisting of spCas9 and single-guide RNA (sgRNA) can recognize target DNA sequences of 22 nucleotides, including specific insertions and NGG motifs, thereby accommodating several nucleotide mismatches. This characteristic implies that particular nuclease complexes may potentially interact with thousands of off-target sites within the human genome. Such off-target activity can result in unintended mutations at non-target genomic loci, as well as chromosomal rearrangements such as translocations, deletions, and inversions. These occurrences pose significant concerns regarding both research and clinical applications of genome editing technologies.
In response to these challenges, various strategies have been devised to mitigate the off-target effects of RGENs. Among these strategies are the addition of two guanine nucleotides to the 5' end of sgRNA, truncation of sgRNA, pairing Cas9 with nickase enzymes, the utilization of catalytically inactive Cas9 (dCas9)-FokI fusion proteins, and the direct delivery of purified Cas9 protein. Empirical evidence demonstrates that such approaches can effectively reduce off-target mutation frequencies by at least an order of magnitude at multiple loci. However, it remains indeterminate whether these enhanced complexes can achieve genome-wide editing that is devoid of off-target activity.
To address this critical issue comprehensively, it is imperative for researchers to develop methods capable of unbiasedly assessing the specificity of these complexes on a genomic scale. In this context, the present study employs whole-genome sequencing (WGS) to evaluate the off-target editing effects of RGENs in cloned gene knockout cells. Additionally, off-target sites were discerned through genome-wide sequencing of nuclease-digested genomic DNA across a substantial number of cells. The results indicate that RGENs manifest high specificity, inducing off-target mutations at a limited number of loci rather than widespread throughout the genome. Furthermore, these off-target effects can be significantly minimized through the optimization of sgRNAs.
The researchers utilized the human haploid cell line HAP1 to generate clonal knockout (KO) cell populations harboring RNA-guided engineered nuclease (RGEN)-induced mutations. The haploid nature of HAP1 cells provides a significant advantage: the sequencing depth for a given number of reads is effectively doubled compared to diploid cells, thereby eliminating the confounding effects of heterozygous variations. Five distinct HAP1 knockout lines were established, each featuring a knockout of a different kinase gene (ABL1, EPHB2, ERBB3, FGFR2, and FGFR4). Genomic DNA was then extracted from these knockout lines as well as from wild-type cells for subsequent whole-genome sequencing (WGS). To validate the reproducibility of the WGS methodology, the research team performed repeat sequencing on both wild-type and ABL1-knockout cells.
Given that RGENs infrequently induce base substitutions, the researchers focused on identifying small insertions and deletions (indels) relative to the hg19 reference genome, employing the Isaac aligner for this purpose. After rigorous filtering, they identified between 2026 and 3250 unique indels across the genomes. A comparative analysis between these indel sites and the RGEN target sites revealed that only 9 to 84 indel sites contained the protospacer adjacent motif (PAM) sequence, with at least 10 sites showing perfect matches to their respective target sequences. Among these identified indels, only one site was confirmed via Sanger sequencing, and this mutation was determined to be a spontaneous occurrence. Concordantly, all five target gene mutations were verified using either the Isaac aligner or the Integrative Genomics Viewer (IGV).
This work underscores the specificity of RGENs in genome editing applications and highlights the importance of employing robust sequence validation techniques in minimizing off-target effects.
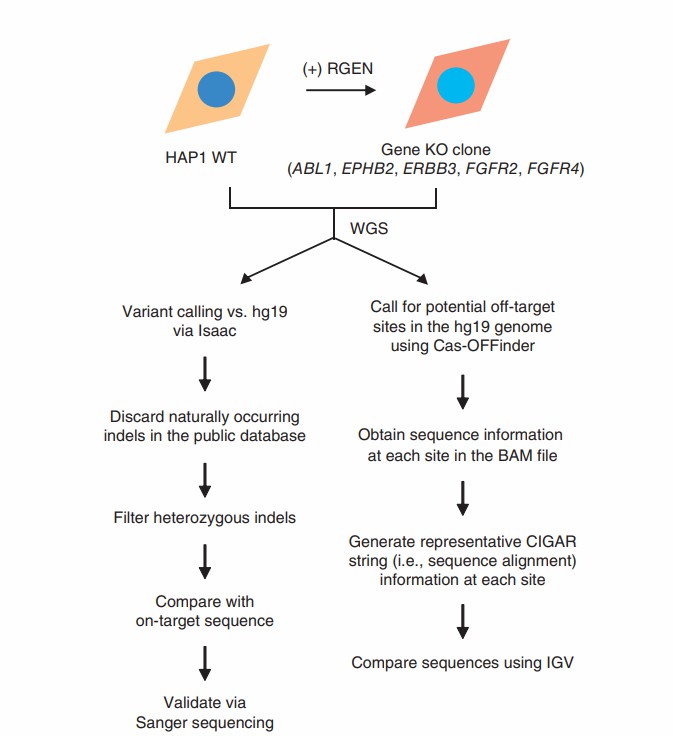 Figure 1. Workflow of off-target analysis of gene KO clones via WGS
Figure 1. Workflow of off-target analysis of gene KO clones via WGS
Subsequently, the researchers utilized Cas-OFFinder and its upgraded version to generate a list of over 100,000 homologous sites with up to 8 nucleotide differences or with DNA or RNA bulge differences of up to 2 nucleotides from the target. These potential off-target sites were then examined for the presence of RGEN-induced indels within each genomic sequence. The research team developed a computational program to compare the read segments surrounding each homologous site with the reference sequence. This program successfully identified all five target mutations in the five knockout clones but did not detect any off-target indels.
The researchers posited that in vitro digestion of the genome by nucleases (digenome sequencing) could identify RGEN-induced off-target mutations within large cell populations. Such digestion products generate numerous DNA fragments with identical 5' ends, resulting in reads that are aligned at the cleavage sites, while unrelated reads exhibit a more interspersed arrangement. The study employed an RGEN specific to the HBB gene, which can induce off-target mutations at highly homologous sites (designated as OT1). Additionally, three other homologous sites (OT3, OT7, and OT12) with 3 nucleotide differences from the target were analyzed. The RGEN effectively cleaved the target, OT1, and OT3 sites both in vitro and within the genomic context.
In digenome samples processed with the HBB-specific RGEN, reads aligned systematically at the target, OT1, and OT3 sites, while no such alignment was observed in digenome samples without RGEN treatment. In contrast, reads spanning breakpoints and exhibiting interspersed patterns were observed at OT7 and OT12.
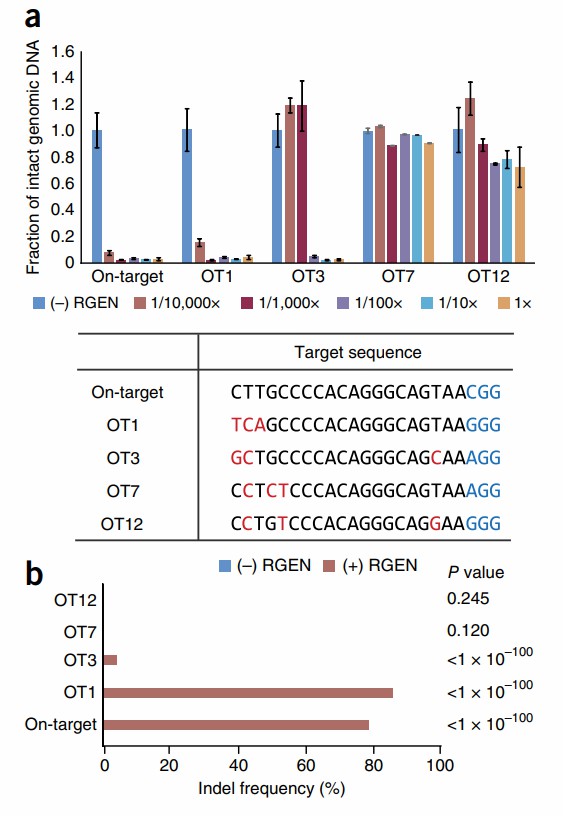 Figure 2. RGEN-mediated genomic DNA digestion in vitro
Figure 2. RGEN-mediated genomic DNA digestion in vitro
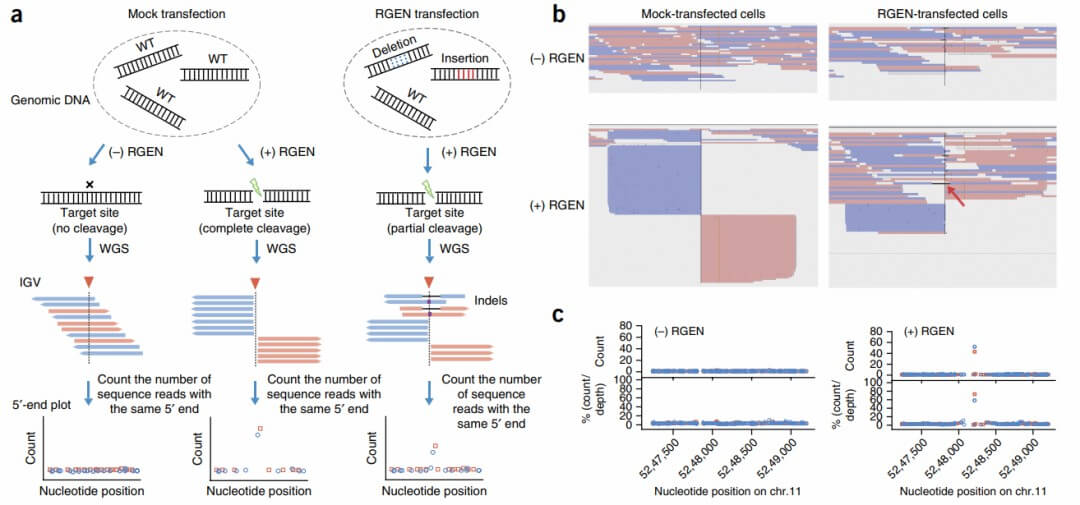 Figure 3. RGEN-induced 'digenome' sequencing to capture off-target sites
Figure 3. RGEN-induced 'digenome' sequencing to capture off-target sites
The research team developed a computational program designed to search for systematically aligned reads across the entire genome. Initially, the researchers mapped the count of sequence reads at single-nucleotide resolution, focusing on nucleotide positions starting from the 5' end near the HBB target site and at two validated off-target sites. The hypothesis posited that an equal number of reads should be present on both the positive and negative strands, adjacent to each other on either side of the cleavage site, resulting in a bimodal distribution. As anticipated, the digenome exhibited bimodal peaks at three cleavage sites. No such bimodal pattern was observed at these sites in genomes not treated with RGEN.
Subsequently, this approach was applied to digenomes derived from RGEN-transfected samples, mock-transfected digenomes, complete genomes treated with RGEN, and complete genomes subjected to mock transfection. Additionally, in vitro treatments were performed using Cas9 protein without sgRNA or with a 100-fold reduction in RGEN concentration (3 nM Cas9) on mock-transfected genomic DNA, followed by whole-genome sequencing (WGS) and digenome analysis. Computational methods were employed to identify sites with more than 10 reads with identical 5' ends on both strands, and those where at least 20% of reads exhibited systematic alignment.
Seventeen and seventy-eight sites were identified in mock digenomes treated with 3 nM and 300 nM RGEN, respectively. These included the target site and two validated off-target sites, all of which displayed bimodal patterns at the 5' end and were arranged linearly in Integrative Genomics Viewer (IGV) images. All identified positions in the complete genomes were false positives, representing naturally occurring indels within the genome. Consequently, the bimodal pattern or systematic alignment of reads was a distinctive feature specific to the three digenomes.
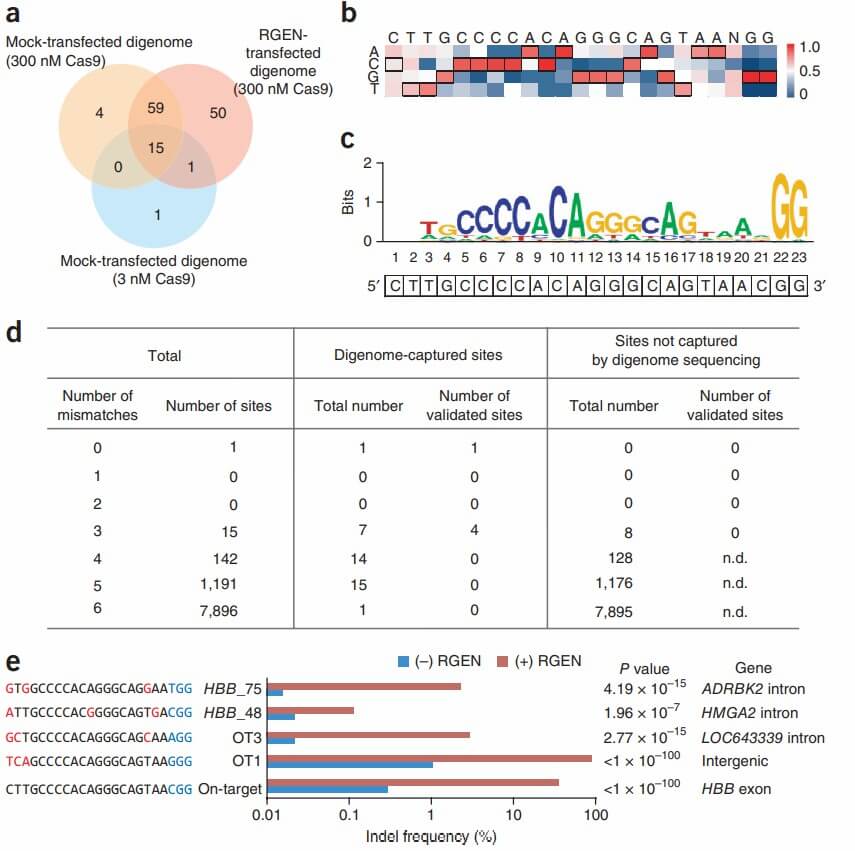 Figure 4. Off-target sites of the HBB RGEN captured by Digenome-seq and validated by targeted deep sequencing
Figure 4. Off-target sites of the HBB RGEN captured by Digenome-seq and validated by targeted deep sequencing
Finally, the research team conducted targeted deep sequencing on 74 common sites identified in two independent digenomes to validate the presence of off-target effects. Additionally, eight other sites, differing by three nucleotides from the target site and not captured by digenome-seq, were tested. No off-target effects were detected at these eight sites, with a frequency of at least 0.1%, which was greater than that observed in negative controls (Fisher's exact test, P < 0.01).
Among the 74 common sites, five (including the verified target site, OT1, and OT3) exhibited indels, with frequencies ranging from 0.11% to 87%. Two newly identified sites, referred to as HBB_48 and HBB_75, had indel detection rates of 0.11% and 2.2%, respectively. These sites differ from the target site by three nucleotides. Compared to the 20-nucleotide sgRNA sequence, the HBB_48 site has three nucleotide mismatches, while the HBB_75 site has two nucleotide mismatches, with a 1-nucleotide difference at the 5' end from the target site. Neither of these validated off-target sites exhibited significant peaks or atypical PAM sequences (5'-NGA-3' or 5'-NAG-3').
These two new off-target sites, along with the other three sites, were independently captured in the three digenomes, demonstrating the high sensitivity and reproducibility of digenome-seq.
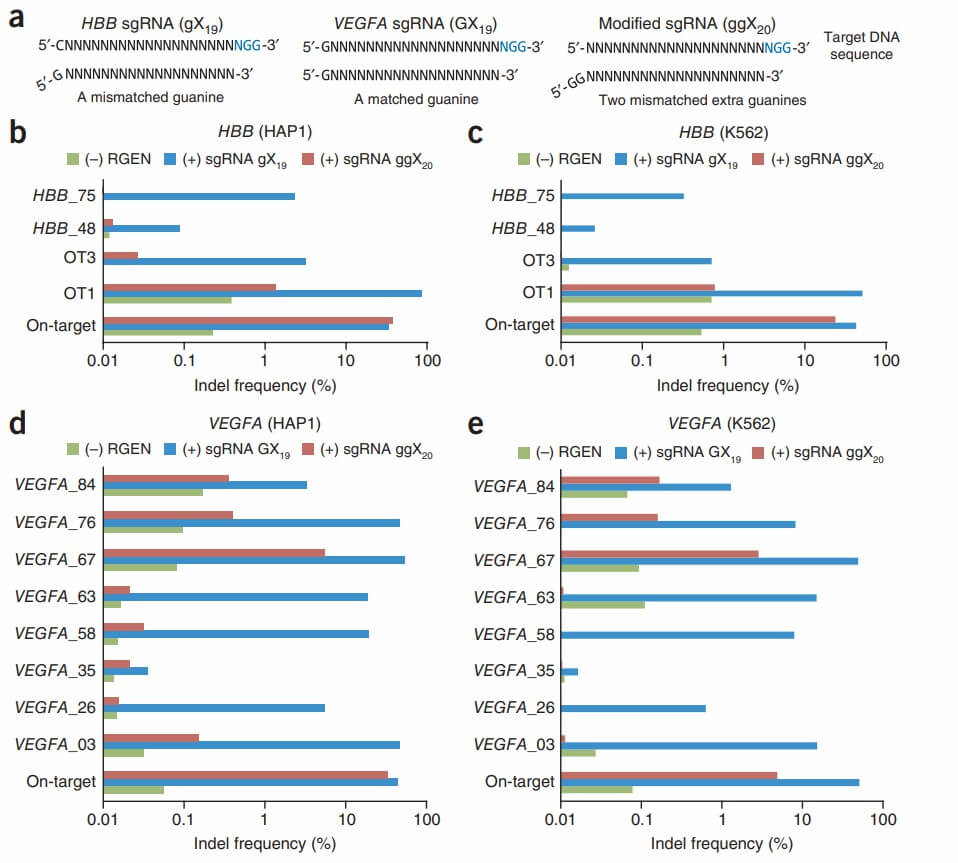 Figure 5. Comparison of conventional sgRNAs with modified sgRNAs that include two extra guanine nucleotides
Figure 5. Comparison of conventional sgRNAs with modified sgRNAs that include two extra guanine nucleotides
CD Genomics is dedicated to advancing research through comprehensive CRISPR/Cas9 off-target detection and analysis services.
Take the Next Step: Explore Related Services
Learn More
Comprehensive Methods for Off-Target Detection in Gene Editing
The genome-wide specificity of the CRISPR-Cas system forms the foundation of RNA-guided genome editing methods. However, data concerning RGEN (RNA-guided endonuclease) specificity appear to be inconsistent. The research team initially employed T7E1 analysis, followed by deep sequencing, which indicated that RGEN does not exhibit off-target effects, even at sites differing by 2 nucleotides (nt) from the target site in human cells. Consistent with these findings, whole-exome sequencing and WGS demonstrated that RGEN did not induce off-target indels throughout the exome and genome in cloned cell populations. In stark contrast, reports from several other research groups suggest that RGEN can induce off-target indels at sites differing by 5 nt or with variations in nucleotide quantity. According to these results, a single RGEN may potentially recognize and cleave thousands of off-target sites within the genome. It is nearly impossible to measure off-target effects at all these potential sites individually. RGEN might induce indels at hundreds or thousands of off-target sites with frequencies below 1% or 0.1%, which are undetectable by sequencing a limited number of clones. Additionally, it is possible that sgRNAs exhibit a broad range of specificities, with some being highly specific and others less so.
To address these issues, the research team selected two sgRNAs with known poor specificity (which have been confirmed to induce high-frequency off-target mutations in human cells) and assessed their genome-wide off-target effects using Digenome-seq. This study demonstrates that Digenome-seq is a reproducible and sensitive method for unbiasedly analyzing RGEN off-target effects.
Firstly, Digenome-seq relies on DNA cleavage rather than binding. Secondly, unlike in vitro selection methods, Digenome-seq operates at the genome-wide scale. Notably, potential off-target sites associated with DNA/RNA bulges can be captured by Digenome-seq. Thirdly, Digenome-seq possesses sufficient sensitivity to detect off-target sites with indel frequencies below 0.1%, approaching the detection limits of high-throughput sequencing platforms. Fourthly, Digenome-seq is cost-effective; with the Illumina HiSeq X Ten, the cost of WGS can now be as low as $1,000 per run. Lastly, the method exhibits good reproducibility. Unlike HTGTS (High-Throughput Genome-wide Transcriptional Screening), Digenome-seq is not constrained by chromatin accessibility. Additionally, HTGTS require the filtration of capture sites with poor homology to the target site, discarding many false positives generated by randomly occurring double-strand breaks (DSBs) or PCR artifacts in the cells. Moreover, the variability in target sequence length due to DSB repair necessitates the search for homologous sites around the capture site, both of which introduce bias.
Reference
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.


