Long-Read Sequencing of DNA Methylation
DNA methylation analysis can help researchers and professionals gain insight into gene regulation as well as identify potential biomarkers. DNA methylation tends to occur in a relatively wide range of genomic regions in both normal and disease conditions. However, the current understanding of DNA methylation is still pieced together from fragmented information obtained by short-read sequencing. Emerging third-generation sequencing technologies (PacBio SMRT and Nanopore sequencing) can directly detect DNA methylation modifications. Using these newer sequencing platforms, CD Genomics is dedicated to providing a comprehensive elucidation of DNA methylation, providing valuable information on gene expression regulation at the chromosomal level.
DNA Methylation Analysis Overview
DNA methylation plays an essential role in epigenetic regulation of cells, such as X-chromosome inactivation, genomic imprinting, transposon suppression, and tumorigenesis, and is one of the hot spots of current research. DNA methylation is a biological process in which methyl groups are covalently incorporated into DNA molecules. 5-Methylcytosine (5mC) is the most common form of this process and is the most studied and best understood modification in plants and animals. N6-methyladenine (6mA), 4-methylcytosine (4mC), and 5mC are frequent in bacteria, having an impact on physiology as well as virulence.
Currently, to detect 5mC, the combination of bisulfite treatment and massively parallel DNA sequencing, namely, bisulfite sequencing (BS-seq) is the most widely used. However, this method has a number of disadvantages, including lack of specificity (inability to distinguish between 5mC and 5-hydroxymethylcytosine, 5hmC), short reads with low sequence diversity, DNA degradation, and the need for specialized protocols. PacBio SMRT sequencing and Oxford Nanopore sequencing are two main third-generation sequencing methods. They allow unambiguous mapping of repetitive and complex regions of the genome and have been widely utilized to detect structural variants, perform phasing haplotypes, and assemble genomes.
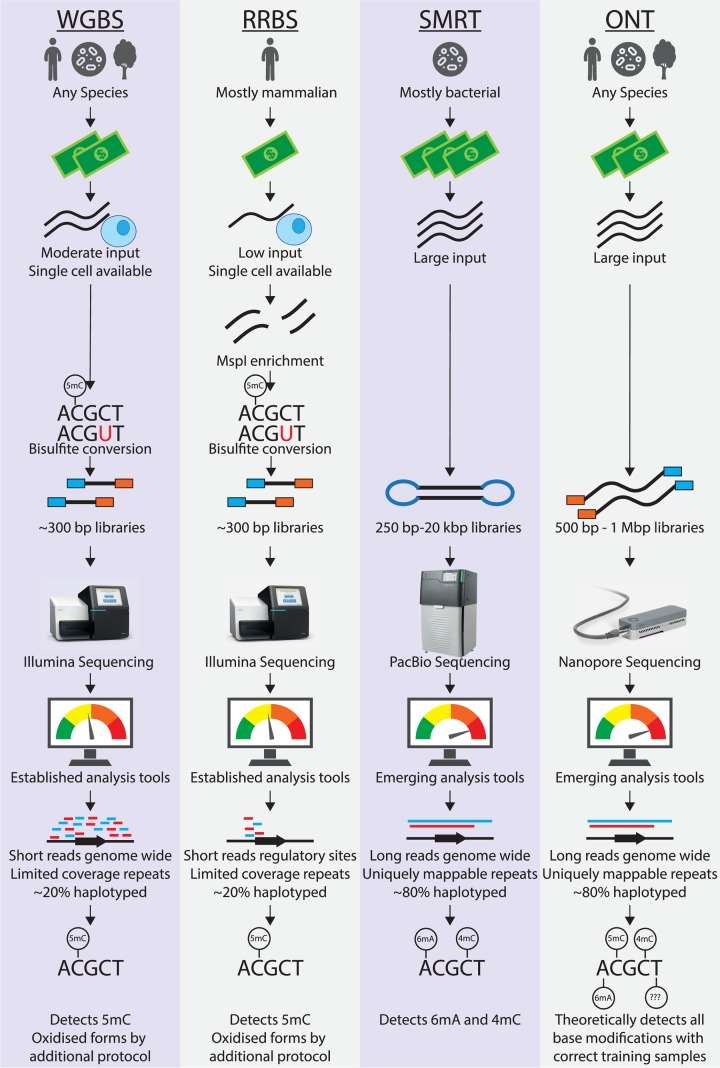 Fig1. Detection of base modifications by bisulfite and long-read sequencing. (Gouil, Q. and Keniry, A., 2019)
Fig1. Detection of base modifications by bisulfite and long-read sequencing. (Gouil, Q. and Keniry, A., 2019)
Long-Read Whole-Genome Analysis of DNA Methylation
SMRT and nanopore long-read sequencing enable the simultaneous detection of a range of base modifications without the need for additional sample preparation. Each method has its own detection focus.
- PacBio SMRT sequencing is most sensitive for 4mC and 6mA detection and is suitable for bacterial epigenomics.
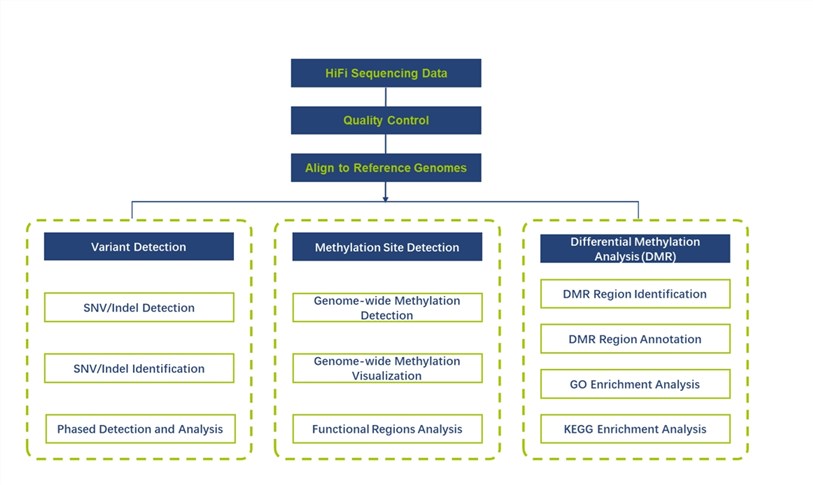 PacBio HiFi sequencing for methylation analysis – CD Genomics
PacBio HiFi sequencing for methylation analysis – CD Genomics
- Oxford Nanopore sequencing is capable of detecting a range of base modifications including 6mA, 5mC, 5hmC, and BrdU, and performs best in detecting 5mC in particular. Compared to SMRT sequencing, nanopore sequencing has a lower cost per Gb and is suitable for larger genomes.
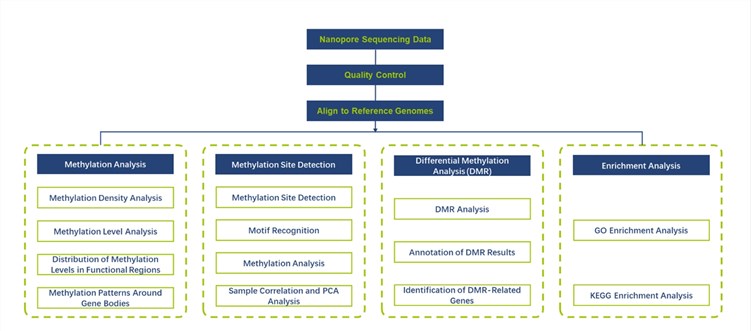 ONT sequencing for methylation analysis – CD Genomics
ONT sequencing for methylation analysis – CD Genomics
Sample Requirements
-Sample type: conventional plant and animal samples.
-DNA amount: ≥ 10 μg, sample concentration ≥ 100 ng/µl.
-Sequencing strategy: non-PCR amplification library construction, Nanopore PromethION and PacBio Sequel ll platform.
Workflow of Our Service

Analysis Contents
- Data re-processing and QC.
- Reference sequence alignment.
- Detection of different types of base modification sites.
- Base modification degree analysis.
- Regional analysis of differential methylation.
- Functional analysis of base modified genes.
DNA methylation, a common epigenetic modification, affects the regulation of gene expression and is involved in a variety of human diseases. DNA methylation is also a common topic in the field of agricultural genomics and can be used to explore responses to drought, extreme temperatures, and other environmental changes. If you are interested in our services, please don't hesitate to contact us. We've got everything covered for your needs and are ready to assist.
Case Study 1: Uncovering Rare Disease Alleles Through 5-base HiFi Genome Sequencing
Background
Rare diseases often pose diagnostic challenges due to their genetic complexity and limited understanding. In such cases, advanced sequencing techniques are essential for identifying causative genetic variants. One such technique is High-Fidelity (HiFi) genome sequencing, which enables accurate detection of single nucleotide variants, indels, structural variants, and, notably, CpG methylation. This case study delves into the application of HiFi sequencing to uncover rare disease-causing alleles through the detection of hypermethylation events.
Methods
A dataset of 276 samples from 152 families, all of which had unresolved rare disease cases, was selected for HiFi genome sequencing. The primary aim was to identify rare hypermethylation events occurring at a frequency of approximately 0.5% within the genome. The sequencing also sought to determine whether these hypermethylation events were allele-specific, as this information could be critical for understanding their impact on regulatory element activity.
Advanced algorithms designed to simultaneously detect CpG methylation within HiFi reads were employed in this study. The team analyzed the resulting data, focusing on the genomic regions exhibiting hypermethylation. The study aimed to establish heritability patterns associated with extreme hypermethylation, categorizing both short (~200 bp) and large (>1 kb) hypermethylation events and identifying potential downstream transcriptional effects.
 Genomic view of 1.5 kb on chromosome 19 at the DMPK locus. (Cheung et al., 2023)
Genomic view of 1.5 kb on chromosome 19 at the DMPK locus. (Cheung et al., 2023)
Results
The analysis of the 5mC HiFi genome sequencing data yielded significant findings:
- Allele-Specific Hypermethylation: It was discovered that 80% of the identified hypermethylation events were allele-specific. This suggests that these events could lead to a loss of regulatory element activity for the affected allele.
- Variants Associated with Hypermethylation: The study identified rare cis variants associated with both short and large hypermethylation events. This is crucial for understanding the genetic underpinnings of these events and their potential clinical significance.
- Repeat Amplifications and Allelic Silencing: Repeat amplifications in proximal promoters were found to predict allelic silencing through hypermethylation. This underscores the importance of regulatory element activity in gene expression regulation.
- Rare Hypermethylation Events Overlapping Disease Genes: On average, each patient exhibited 30-40 rare hypermethylated genomic regions overlapping with rare disease genes. This finding suggests a potential link between hypermethylation and rare diseases.
- Identification of a Causative Allele: The study resulted in the identification of a previously undiagnosed causative allele, specifically DIP2B, responsible for overall developmental delay. This underscores the potential of HiFi genome sequencing to uncover unconventional disease alleles linked to the loss of regulatory element activity.
Case Study 2: 6mA Profiles of Chromatin and Transcription by SMRT Sequencing and NGS
Background
DNA methylation plays a pivotal role in regulating gene expression and chromatin structure in various organisms. One specific form of DNA methylation, DNA N6-adenine methylation (6mA), has been recently discovered in a wide range of eukaryotes, from unicellular organisms to metazoa. Understanding the molecular mechanisms behind 6mA and its impact on chromatin organization is crucial for unraveling its functional significance. In this case study, they delve into the discovery of a DNA 6mA methyltransferase complex in ciliates, known as MTA1c, and its effects on chromatin structure.
Methods
Researchers embarked on a comprehensive investigation to understand the DNA 6mA methyltransferase complex and its role in chromatin organization using MNase sequencing (MNase-seq), poly(A)+ RNA sequencing (RNA-seq), transcriptional start site sequencing (TSS-seq), and single-molecule real-time sequencing (SMRT-seq).
References
- Gouil, Q., & Keniry, A. (2019). "Latest techniques to study DNA methylation." Essays in biochemistry, 63(6), 639-648.
- Tse, O. O., et al. (2021). "Genome-wide detection of cytosine methylation by single molecule real-time sequencing." Proceedings of the National Academy of Sciences, 118(5).
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment