

We are dedicated to providing outstanding customer service and being reachable at all times.

Bacterial Whole-Genome De Novo Sequencing
CD Genomics is providing long-read sequencing technologies developed by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) to fully support the de novo assembly of bacterial genomes. To ensure maximum assembly accuracy, we recommend using short-read Illumina data to further polish the genome assembly. Our services are end-to-end and customized, enabling the rapid delivery of more cost-effective strategies and high-quality genome assemblies.
Using Long-Read Sequencing to Improve De Novo Assembly of Bacterial Genomes
Short-read sequencing technologies, such as the Illumina platform, are now widely used in bacterial genome research due to its ability to provide rapid and accurate analysis of the entire bacterial genome. However, short-read sequencing, in addition to being limited by amplification and sequencing bias, has inherent limitations that generally do not allow for whole-genome assembly. Long-read sequencing technologies developed by ONT and PacBio can produce long reads in tens of kilobase pairs, thus overcoming the limitations of short-read sequencing to obtain a complete assembly. As the first established long-read technology, PacBio Single Molecular Real-Time (SMRT) sequencing is based on a sequencing-by-synthesis method. The PacBio sequencer can generate the average length of reads ranging from 10-14 kb and can reach throughputs of about 30 Gb per run. ONT long-read sequencing has no upper read length limit and can assemble the complete microbial genome relatively quickly and inexpensively, which has gained increasing popularity. This technique has been used to assist in the completion of the genome sequence of Staphylococcus aureus. With the advanced technologies and improved analysis pipelines, ONT long-read sequencing can be applied for complex bacteria, such as Fusobacterium nucleatum, Pseudomonas koreensis, Mycobacterium tuberculosis and other genomes with long repetitive sequences.
Service Highlights
- Years of experience in microbial de novo sequencing projects, including bacterial genomes.
- Facilitating high-quality genome reconstruction with the largest short and long-read sequencing capacity.
- Providing complete and customized solutions for in-depth analysis of bacterial genomes.
- Generation accurate reference sequences, even for complicated bacterial genomes.
Bacterial Whole-Genome De Novo Sequencing Service Overview
| Sequencing Platform | Illumina HiSeq PE150 | PacBio Sequel ll | Nanopore PromethION |
| Sample Requirements | Sample Type: genomic DNA Amount:≥ 200ng OD260/280=1.8~2.0 no degradation and no contamination |
Sample Type: HMW genomic DNA Amount:≥ 15μg OD260/280=1.7~2.0 Fragments should be ≥ 20 kb |
Sample Type: HMW genomic DNA Amount:≥ 10μg OD260/280=1.75~2.0 Fragments should be ≥ 20 kb |
| Sequencing Strategies | 350 bp insert library, ≥ 50 X | 20Kb SMRTbell library, CLR sequencing, ≥100X | ONT Library preparation, ≥100X |
| Application Scope | Large-scale sequencing and rapid scanning | Sequencing of a small number of strains Accuracy in place |
Sequencing of a small number of strains Accuracy in place |
| Delivery Cycle | Minimum of 30 working days | Minimum of 45 working days | Minimum of 40 working days |
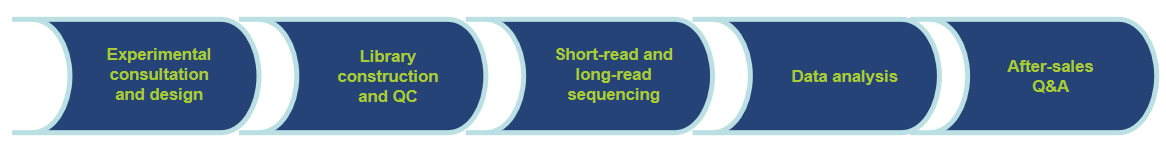
Workflow of Our Services
Analysis Contents
Analysis of bacterial genome information at different levels of fineness, deciphering the development of bacteria, and exploring their potential value. Our analysis consists of two main parts: standard analysis and advanced analysis. Specific analytical services are as follows, but are not limited to,
- Standard analysis
-Data quality control.
-Genome assembly and evaluation of assembly results.
-Genome component analysis.
-Gene functional annotation.
-Genome cycle graph generation. - Advanced analysis
-Comparative genome analysis.
-Genome island prediction.
-Prophage prediction.
-Bacterial virulence-related gene annotation.
-Bacterial drug-resistance gene annotation.
-Phylogenetic analysis.
To perform bacterial whole-genome de novo sequencing, researchers choose CD Genomics. With multiple specialists and years of experience in this field, we guarantee you high-quality data and integrated bioinformatics analyses. Please feel free to contact us. We are happy to help you!
FAQ
Can you provide a completion map of a bacterial genome?
Absolutely. We can create a comprehensive mapping of a bacterial genome for you. Bacterial genomes possess distinctive characteristics that set them apart from other species:
- Compact Size: Bacterial genomes are typically small, often ranging from less than 10 million base pairs (Mbp) with the majority falling in the 4 to 5 Mbp range.
- Single Chromosome: Most bacterial species have a single circular chromosome as their primary genetic material. In addition to this, some bacteria may also contain plasmid sequences, which are smaller, circular pieces of DNA.
- Variable GC Content: Bacterial genomes exhibit a wide range of GC content, varying from as low as 25% to as high as 70% or more. This GC content plays a crucial role in genome stability and adaptation.
- Simplicity in Composition: Bacterial genomes are relatively straightforward in composition. Typically, over 80% of the genome consists of coding regions, which means that genes constitute a substantial portion of the genome. Moreover, bacterial genomes tend to have fewer repetitive sequences compared to more complex organisms.
- Gene Continuity: Bacterial genes are generally contiguous, lacking introns. This continuous arrangement simplifies the process of identifying genes and their functions.
What is the meaning of "detection of mobile elements and resistance genes" in my bacterial genome report?
These two categories are both part of genome matching, mobile elements are transposons and so on, and resistance genes are specific genes. Mobile elements refer to a type of DNA that can change its position within a genomic region, including transposons, plasmids, phage elements, etc. Resistance genes refer to genes with a particular resistance, such as antibiotic resistance genes.
How to ensure zero gap for the bacterial genome completion map and what requirements do the samples need to meet?
Zero gaps typically entail a single contig assembly, while a high-quality finished map should ideally comprise no more than three contigs. However, successful genome completion may be compromised under the following conditions:
- Bacterial genome size exceeding 6 million base pairs.
- Presence of impurities in the sample, including but not limited to sequence contamination from other species.
- Cumulative count of chromosomal genomes and plasmids equaling or surpassing five.
- Existence of transposons or other repetitive sequences exceeding 20,000 base pairs, or an exceptionally high proportion of such sequences in the genome, exceeding 20%.
- Sample DNA quality falling below the standards required for long-read sequencing, with varying gap tolerances for different sample grades.
- A GC content greater than 70% or less than 30%.
What sets microbial completion maps apart from microbial genome sequencing?
Microbial resequencing entails variant detection through high-throughput sequencing and comparison with a closely related reference genome. This method yields a range of variant data, such as single nucleotide polymorphisms (SNPs), insertions/deletions (InDels), structural variations (SVs), and more. These variants can be dissected to understand trait disparities between genomes or employed as markers for comprehensive evolutionary analysis.
Distinctions between fungal/bacterial framework maps, fungal/bacterial fine maps, and bacterial completion maps primarily revolve around assembly completeness and sequencing strategies. The bacterial completion map utilizes PacBio sequencing, tailoring the strategy to the specific characteristics of the bacterial strain in question. This approach aims for a fully finished genome sequence, characterized by a single scaffold with minimal gaps (ideally ≤5 gaps). It represents the highest standard for bacterial genome sequencing assembly.
Which structural annotations are encompassed within the bacterial completion map?
This includes predictions for various genetic elements such as genes (gene, CDS, tRNA, rRNA, 23S rRNA, 16S rRNA, 5S rRNA, miscRNA, tmRNA), pseudogenes, CRISPR sequences, gene islands, prophages, repetitive sequences, and secondary metabolism gene clusters. For more customized report needs, you can contact us.
Which databases contribute to the functional annotation within the bacterial completion chart?
This encompasses both widely used general-purpose databases (such as Nr, KEGG, GO, Swiss-Prot, COG, CARD, CAZy) and specialized proprietary databases (including VFDB, PHI, Antismash, TCDB, ARDB, Pfam, and CYP450).
Is it possible to conduct both the bacterial whole genome sequencing and bacterial methylation analysis together?
Certainly, the experimental workflow for the bacterial completion map and bacterial methylation is identical, allowing for the sharing of data. However, it's important to note that a larger data volume is necessary, and it's recommended to achieve a coverage of 300x or more.
What should be done in the event of strain contamination?
It is advisable to conduct an initial round of strain identification to ensure the presence of a single bacterium before proceeding with subsequent experiments and analyses. In the unfortunate event of contamination being detected after completing all experiments, one possible approach is to increase the data volume to reach the desired depth for the target strain. Nonetheless, it is strongly recommended to prevent contamination from occurring at the outset of the experiment. Strain contamination typically falls into two categories: intraspecies and interspecies contamination. To clarify these concepts:
- Interspecies contamination: This involves interactions between populations of different species, indicating differences in species classification.
- Intraspecies contamination: This refers to interactions between individuals of the same species, signifying that they belong to the same species.
How does sequencing distinguish between genomes and plasmids?
A plasmid is a distinct genetic unit separate from the chromosomal DNA, capable of autonomous replication. It encompasses relatively simple deoxyribonucleic acid (DNA) molecules found outside the chromosomes in various organisms, including symbiotic organisms, eukaryotic organelles, and bacterial cells. The study of plasmids encompasses vital theoretical concepts such as genetic recombination, DNA replication, and biological evolution. Furthermore, the potential for drug-resistant plasmids to disseminate among disease-causing bacteria has elevated plasmid research to an epidemiological concern.
Upon assembly, there is a noticeable disparity in size between plasmids and genomes. Additionally, when subjected to comparison against the NR (non-redundant) database, significant distinctions become apparent. Plasmids have the capacity to encode genes and consist of DNA sequences. However, plasmids, in general, do not appear to be thoroughly validated.
References
- Zhang, P., et al. (2021). Comparison of de novo assembly strategies for bacterial genomes. International Journal of Molecular Sciences, 22(14), 7668.
- Liao, Y. C., et al. (2019). Completing circular bacterial genomes with assembly complexity by using a sampling strategy from a single MinION run with barcoding. Frontiers in microbiology, 2068.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment