
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Haplotype is defined as the sequence of genetic variation events on the same chromosome in a cell. It has been established that different genetic variants on the same chromosome are usually transmitted together to the daughter cells, and this chained feature is closely related to the organism's inheritance pattern, allele-specific gene expression, drug sensitivity, and tumour susceptibility. The successful assembly of the first telomere-to-telomere complete human reference genome (T2T-CHM13) represents a significant milestone in the field of human reference genome assembly. The forthcoming era of complete genome research will centre on diploid reference genomes that are fully characterised by their haplotypes (phased), with linkage information being critical for a comprehensive understanding of genetic diversity and its relationship to genetic diseases.
Recent years have seen a paradigm shift in the field of molecular biology with the advent of single-molecule sequencing technologies. These technologies, exemplified by PacBio sequencing and Oxford Nanopore Technologies (ONT) sequencing, have enabled researchers to routinely obtain sequencing read lengths of up to 10 kb. Notably, ONT's Ultra-long technology has demonstrated the capability to achieve N50 values exceeding 100 kilobases, marking a significant advancement in the field. Nevertheless, due to the benefit of extended read lengths inherent to single molecule sequencing, it is feasible to undertake local haplotyping of genetic variants in the genome. However, it is virtually impossible to span mitoplasmas (which are generally in the millions of base pairs in length) and a number of other intricate regions of the genome.
Presently, researchers are combining single-molecule sequencing with a variety of other sequencing technologies in order to achieve haplotype typing at the whole chromosome level, without reliance on parental data. For instance, the integration of PacBio's HiFi data with Hi-C data, which detects the three-dimensional structure of the genome, and the utilisation of remote interactions within the same chromosome to ascertain the linkage information of distant heterozygous single-nucleotide polymorphisms (SNPs) is a method that demands a high degree of accuracy in long-read length sequencing data. It should be noted that some of the highly complex genomic regions involved in the longer structural variation (SV) are subject to typing errors. The second approach involves the integration of long-read length sequencing data from a substantial number of cells with Strand-seq single-cell genomic DNA template strand interlocking sequencing data. Through this process, Strand-seq performs retention strand-specific genomic sequencing of biparental DNA template strands within a single cell. This enables the capture of interlocking information on the same single strand of DNA from the same chromosome. The subsequent process involves the achievement of whole chromosome-level Haplotyping.
However, Strand-seq technology is unable to assist the HiFi long read length sequencing data of a large number of cells for haplotype typing in some complex genomic regions, especially in the absence of heterozygous SNPs in the neighbourhood, and it is unable to type heterozygous structural variants. Strand-seq technology is based on a second-generation sequencing platform with short read lengths and low genome coverage. The Strand-seq technology has been enhanced (OP-Strand-seq) to increase genome coverage to 13%, yet this is contingent on the utilisation of bespoke instrumentation and does not address the genome matching issue inherent to short reads and long sequencing.
Take the Next Step: Explore Related Services
In order to address the aforementioned challenges, Fuzhuang Tang's research group at Peking University's Center for Frontier Innovation in Biomedicine/Champing Laboratory/Peking University-Tsinghua Joint Center for Life Sciences published a research paper in Cell Discovery entitled 'Simultaneous de novo calling and phasing of genetic variants at chromosome-scale using NanoStrand-seq'.
This research was pioneering in its development of a method for chain sequencing of DNA template strands using a single-molecule sequencing platform, termed NanoStrand-seq. The method has been demonstrated to possess the capability for precise ab initio detection and haplotype typing of SNPs and SVs at the whole chromosome level, exhibiting a high degree of typing accuracy (99.98% and 99.68%, respectively). Its applicability extends to both long-term passaged cultured cells and short-term primary cultured cells, thereby satisfying a diverse array of experimental requirements.
In experimental settings, NanoStrand-seq employs 5-bromo-2-deoxyuridine (BrdU, 5′-bromo-2′-deoxyuridine) to label newly synthesised DNA single strands during DNA replication. This process occurs during cell preparation, and subsequent steps in the experimental procedure are undertaken only after the cells have completed mitosis. The NanoStrand-seq methodology is distinct from Strand-seq in its utilisation of Tn5 transposase, a low-concentration enzyme, to fragment single-cell genomic DNA. This approach enhances both the DNA fragment length and the experimental robustness. Subsequent to the dissociation of the Tn5 transposase from the DNA fragments, a gap-filling reaction is performed. Subsequent to this, UV light is utilised to selectively remove the nascent DNA single strand, leaving only the template DNA strand. The method incorporates two rounds of primer extension reactions, adding distinct barcode sequences to both ends of the DNA molecules. By combining these barcodes, both single-cell information and DNA strand-specific information are preserved.Finally, the long DNA fragments undergo PCR amplification and library construction for single-molecule sequencing.
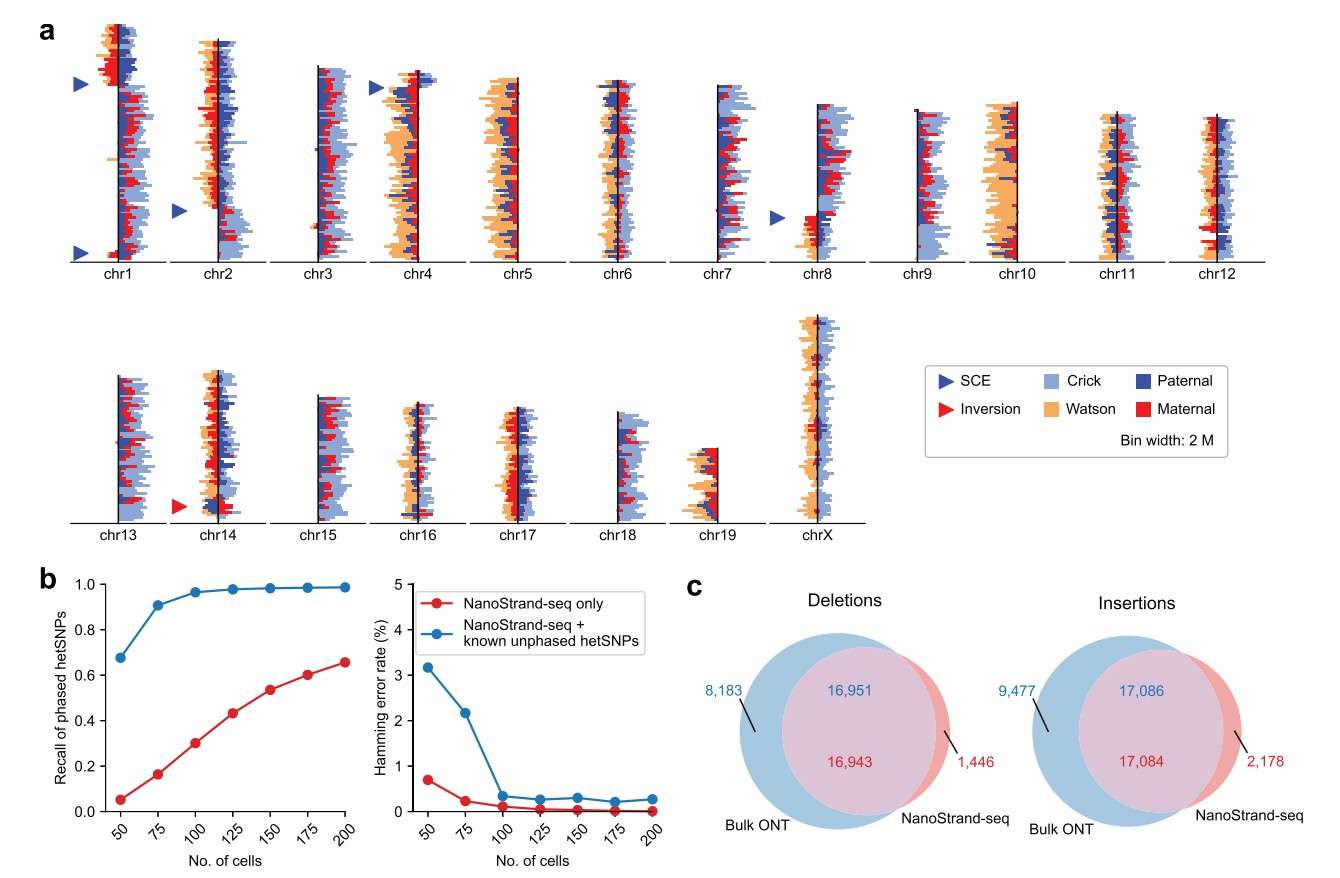
In order to assess the reliability of the NanoStrand-seq technology, the research team applied NanoStrand-seq to the human normal diploid GM12878 cell line and conducted a comprehensive comparison with the Strand-seq/OP-Strand-seq datasets based on next-generation sequencing platforms. The results of this systematic comparison revealed that NanoStrand-seq achieved an average read length of 2,794 bp, with 364 out of 826 cells passing quality control, resulting in a pass rate of 44%, which is significantly higher than the 11% pass rate of the OP-Strand-seq technology. Additionally, the background noise of NanoStrand-seq data (0.88%) is lower than that of Strand-seq (1.40%), and the single-cell genomic coverage (9.39%) is higher than Strand-seq (3.51%). The GC bias of NanoStrand-seq is also closer to the true genomic distribution compared to both Strand-seq and OP-Strand-seq. Furthermore, the NanoStrand-seq data allows for the direct detection of strand-specific distribution, sister chromatid exchange (SCE) events, and genomic inversion events. The DNA reads from the two parental genomes in the same single cell are successfully distinguished based on strand specificity.
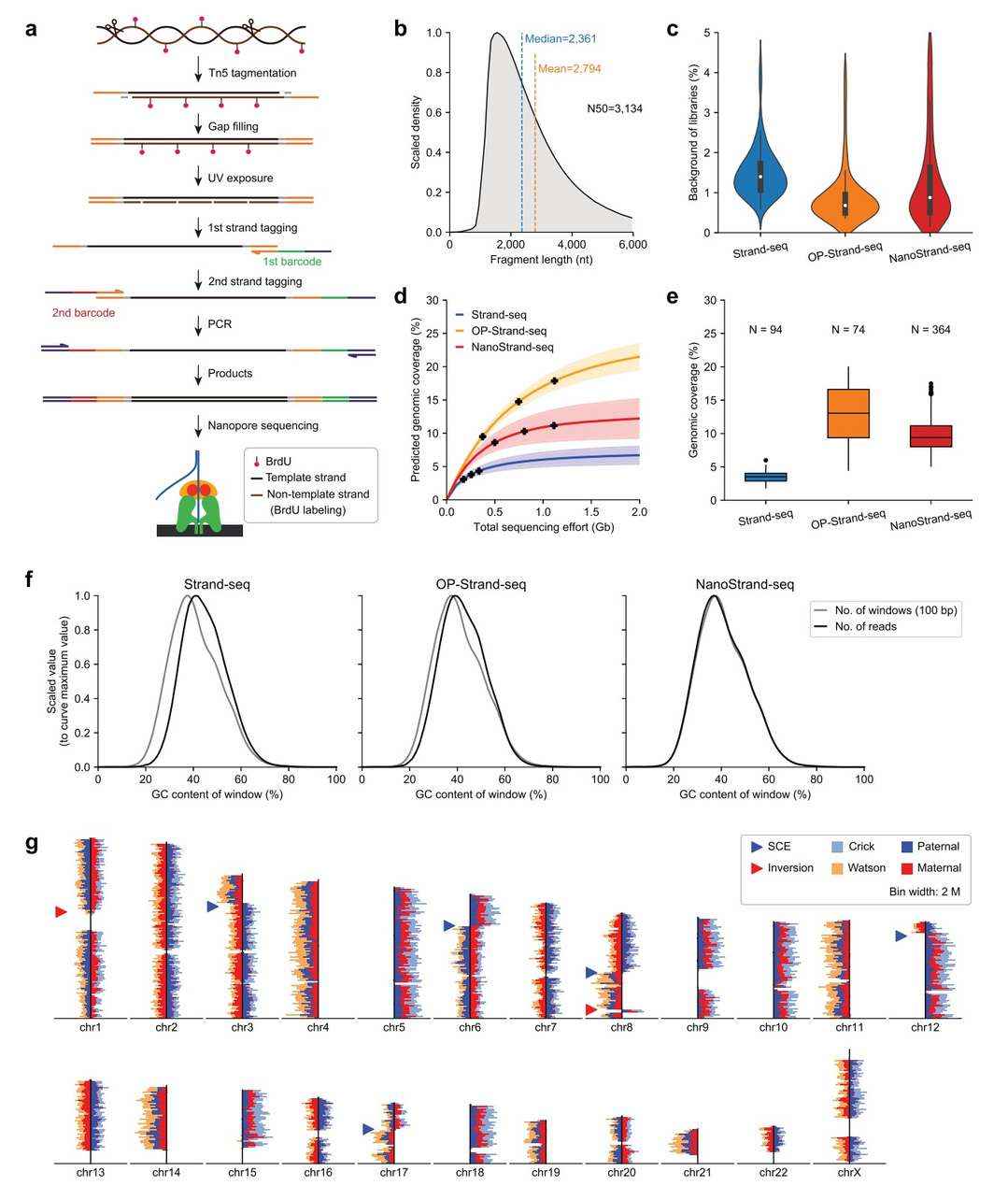 Strategy and characterization of NanoStrand-seq (Bai et al., 2024)
Strategy and characterization of NanoStrand-seq (Bai et al., 2024)
NanoStrand-seq enables quasi-detection of inversion events in genomes
The conventional approach to detecting inversion events, predicated on genome comparison breakpoints, exacts stringent requirements for sequencing depth. The sequences at the breakpoints of inversion events are intricate, and sequence comparison is susceptible to inaccuracies, thus exhibiting significant limitations. In the present study, an algorithm was developed for the detection of genomic inverted regions. This was based on the change of read segment orientation in response to the characteristics of long read segments and the small number of read segments in NanoStrand-seq data. As a result of this research, 339 potential inverted regions (multiple inverted regions may belong to the same complex inversion event) were successfully identified in GM. The study identified 12,878 cells, including a pure inversion measuring 3.89 Mb on chromosome 8 and a 27.43 Kb heterozygous inversion on chromosome 7.
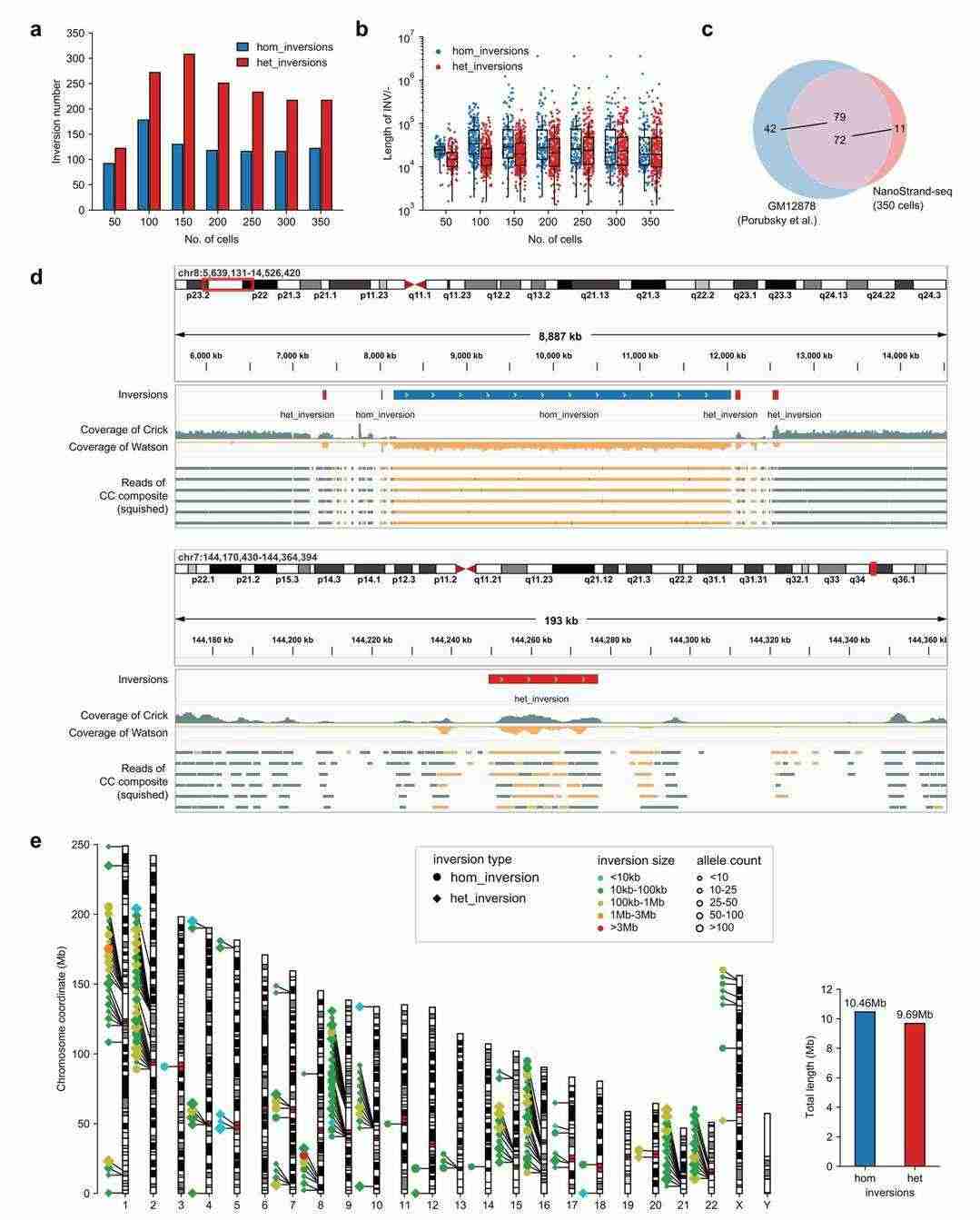 Inversions detected by NanoStrand-seq (Bai et al., 2024)
Inversions detected by NanoStrand-seq (Bai et al., 2024)
Despite its limitations in detecting short inversion events due to the number of read segments, NanoStrand-seq can accurately identify such events through a comparison of breakpoints, utilising the long read length to enhance the recall of inversion event detection. The detected inversion events were predominantly concentrated near the mitre and telomere regions. Furthermore, the total length of the pure inversion region was determined to be 10.46 Mb, while the total length of the heterozygous inversion region was found to be 9.69 Mb.
It is acknowledged that inversion events in the genome are complex. Following a rigorous process of manual filtering, merging and correction, the final 83 inversions were compared with the 121 inversions in the reference set. The analysis revealed that 72 (87%) of the inversions overlapped with 79 (65%) of the inversions in the reference set.
NanoStrand-seq enables accurate ab initio detection and haplotyping of SNPs at the whole chromosome level
Due to the lower accuracy of single-molecule sequencing in comparison with second-generation sequencing, errors can more readily occur during the detection of SNPs, particularly in single-cell data where the sequencing depth is low and a single read cannot accurately determine the genotype of the allele. This study developed an error-tolerant analysis process to address this problem. Firstly, the sequencing data from multiple single cells were directly combined to detect heterozygous SNPs from scratch. Then, PCR repeats in the single-cell data were used to correct single-base sequencing errors, and these heter Heterozygous SNPs were utilised as anchors to cluster the cells into two classes and subsequently merge them separately to obtain the haplotype reads (in terms of the whole chromosome). Finally, the allele genotypes (in terms of whole chromosomes) were determined based on the multiple cells in a supported manner.
It is important to note that this process is the first to introduce a strategy of two rounds of read segment typing. The first round uses the strand-specific features of single-cell reads to extract haplotype reads. The second round labels single-cell reads with the typed SNPs from the first round. This re-extracts the haplotype reads. This improves the accuracy of the haplotype typing of the reads. It also improves the recall and accuracy of the SNP typing. The process outputs SNPs with typing information that are further corrected for identified pure inversion events.
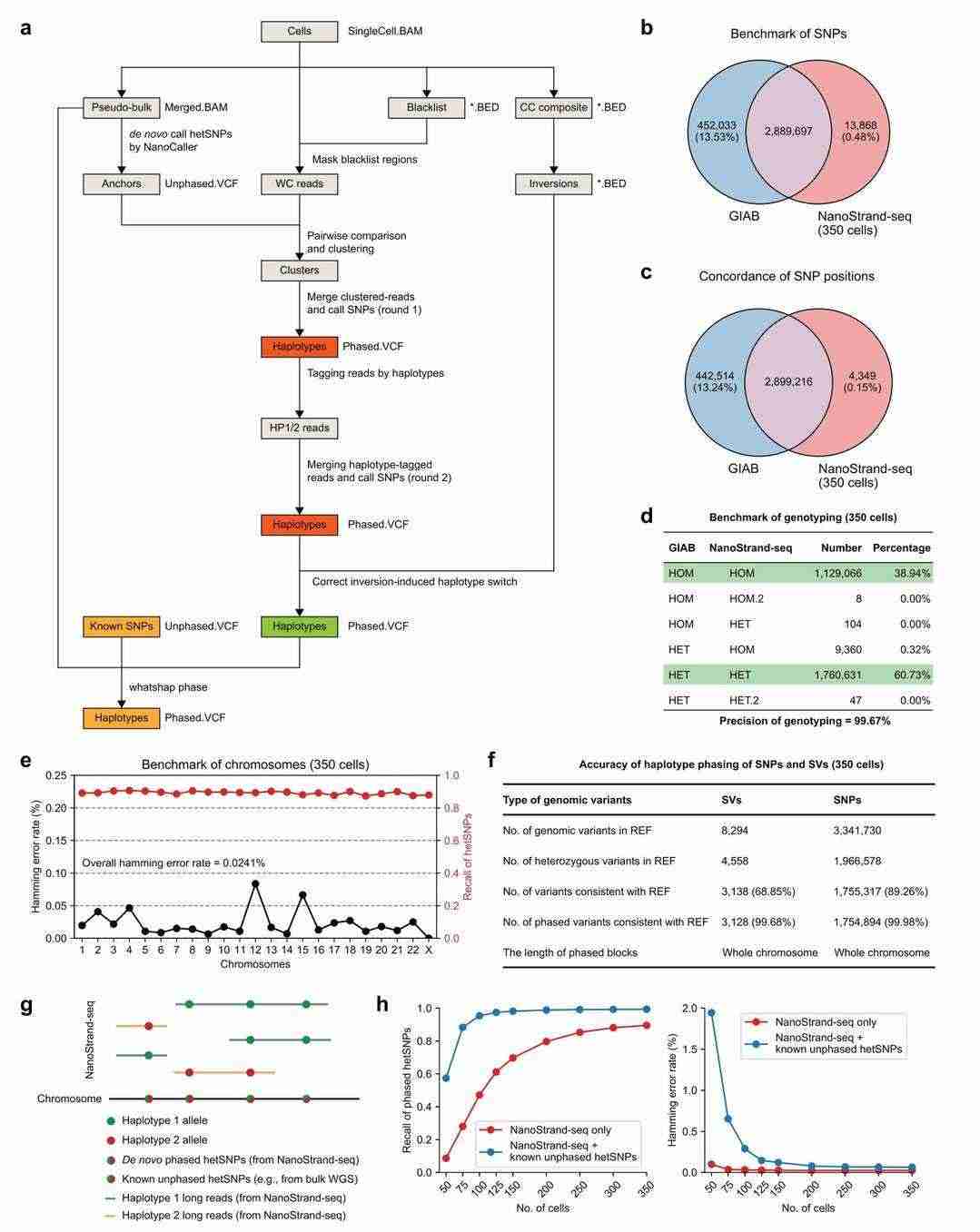 The strategy of de novo calling and phasing of hetSNPs across the whole genome (Bai et al., 2024)
The strategy of de novo calling and phasing of hetSNPs across the whole genome (Bai et al., 2024)
A comparison with the SNP gold standard set in GIAB demonstrated that NanoStrand-seq detected SNP locations with 99.85% accuracy, 86.76% recall, and 99.67% accuracy in further determining the genotype using sequencing. The data set comprised 350 single cells, and 89.26% (1,755,317) of heterozygous SNPs in GIAB were successfully detected by NanoStrand-seq. The accuracy of haplotype typing was 99.98%.
Moreover, the study analysed the capacity to type 95.35% of heterozygous SNPs with 99.71% typing accuracy utilising a mere 100 single cells, and 99.33% recall with 99.94% typing accuracy employing 350 cells, under circumstances where the location and genotype of the heterozygous SNPs were known, yet the typing information was unavailable.
NanoStrand-seq enables precise haplotype typing of SNPs in highly complex genomic regions such as the major histocompatibility complex MHC
It has been demonstrated that long read lengths are conducive to the generation of more accurate comparison results for short read lengths in complex genomic regions.This study further evaluated the advantages of NanoStrand-seq for detecting and typing SNPs in the major histocompatibility complex (MHC) region. The MHC region is characterised by significant heterogeneity, spanning approximately six million base pairs (6Mb) within the human genome. It is noteworthy that substantial discrepancies exist in the annotation of this genomic region, even when comparisons are made between the gold standards of the GIAB and the 1000 Genomes Project.
A simultaneous comparison was made with GIAB and 1000 Genomes, with 16,731 SNPs common to all three datasets. The resultant genotypes were determined with 99.55% accuracy and haplotyping with 100% accuracy when compared to GIAB, and with 98.8% accuracy and haplotyping with 98.93% accuracy when compared to 1000 Genomes.
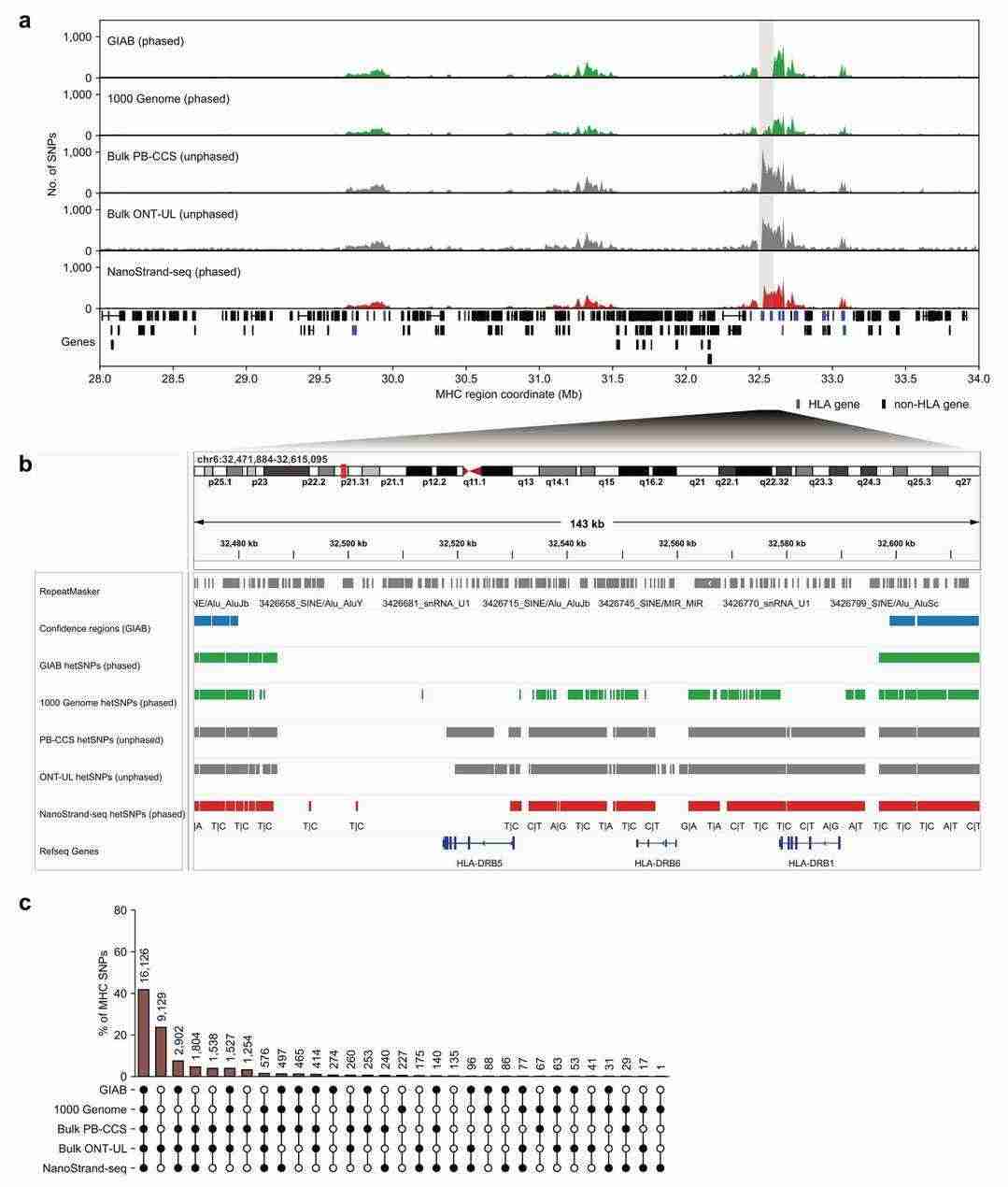 The performance of NanoStrand-seq in calling SNPs within high-complexity genomic regions (Bai et al., 2024)
The performance of NanoStrand-seq in calling SNPs within high-complexity genomic regions (Bai et al., 2024)
It is important to note that NanoStrand-seq was able to detect a greater number of SNP sites at the human leucocyte antigen (HLA) genes in the MHC region when compared to GIAB and 1000 Genomes. Furthermore, the bulk cell ONT Ultra-long data and bulk cell PacBio HiFi data demonstrate comparable SNP densities to NanoStrand-seq. However, it should be noted that these data lack information regarding haplotype typing at the whole chromosome level. A comparison of the NanoStrand-seq data with the PacBio HiFi data for a large number of cells revealed that 97.31% (22,314/22,932) of the SNP positions were consistent with the results from the HiFi sequencing data, with up to 99% accuracy in determining the genotype.
NanoStrand-seq enables precise ab initio detection and haplotyping of structural variants at the whole chromosome level
The study combined single-cell reads and detected 21,211 structural variants, including insertions and deletions. These structural variants were then characterised using haplotype reads, resulting in the successful typing of 3,322 heterozygous deletion events and 2,688 heterozygous deletion events. In order to assess the accuracy of structural variation detection, this study constructed a structural variation reference set based on HiFi data and Ultra-long data, and generated a blacklist of comparison regions. The results of the comparison of the structural variants with HiFi show that the recall and accuracy of missing events are 91.99% and 83.6%, respectively, and the recall and accuracy of insertion events are 86.09% and 87.01%, respectively.
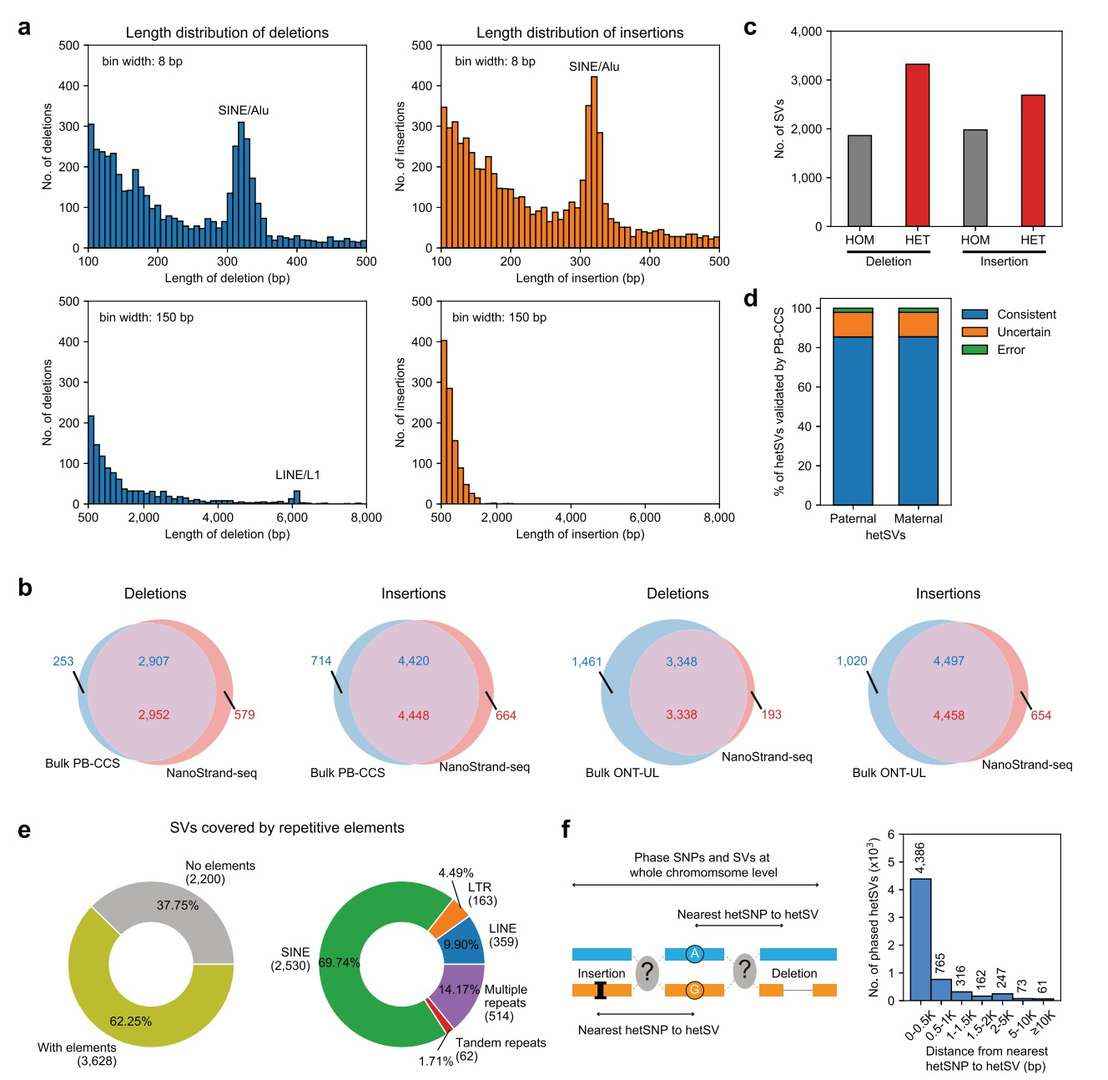 The performance of NanoStrand-seq in detecting and phasing SVs (Bai et al., 2024)
The performance of NanoStrand-seq in detecting and phasing SVs (Bai et al., 2024)
For the heterozygous structural variants successfully typed by NanoStrand-seq, 87.61% were verified by HiFi data and GIAB (97.61% accuracy), and 12.42% of the structural variants exceeded the ability to be verified by the HiFi data, demonstrating the superiority of NanoStrand-seq in the haplotype typing of these structural variants. Furthermore, utilising GIAB for the typing of the aforementioned structural variants in HiFi, a total of 3,138 heterozygous structural variants were detected by NanoStrand-seq, thereby demonstrating an accuracy of 99.68% in terms of haplotype typing.
Strand-seq, a second-generation sequencing platform, has also been shown to be capable of typing structural variants with long read-length data. However, this is dependent on the presence of heterozygous SNPs in the vicinity of the structural variants in question. From a statistical perspective, A total of 381 heterozygous structural variants were found to have distances to the nearest heterozygous SNP of more than 2 Kb, and 61 were found to have distances to the nearest heterozygous SNP of more than 10 Kb. The present study validated nine of these structural variants by targeted single-molecule sequencing, and the results were all consistent with the typing results from NanoStrand-seq. However, neither the HiFi data nor the Ultra-long data were able to determine haplotype typing information for these heterozygous structural variants.
NanoStrand-seq can guide the de novo assembly of haplotype typing genomes
In this study, the human genome was assembled from scratch using a substantial amount of cellular HiFi data to generate multiple contig clusters. These contig clusters were then clustered and directionally corrected with the assistance of NanoStrand-seq data, a process which ultimately enabled the accurate clustering of contig clusters. From the same chromosome into the same class, with a high accuracy of 99.35% in terms of length, and then detecting and typing the SNP loci in the contigs, and mapping the SNPs in the clusters back to the hg38 reference genome. Subsequently, the SNP sites in these clusters were detected and typed, and following the mapping of the SNPs in the clusters back to the hg38 reference genome, the typing accuracy was assessed to be 99.66%.
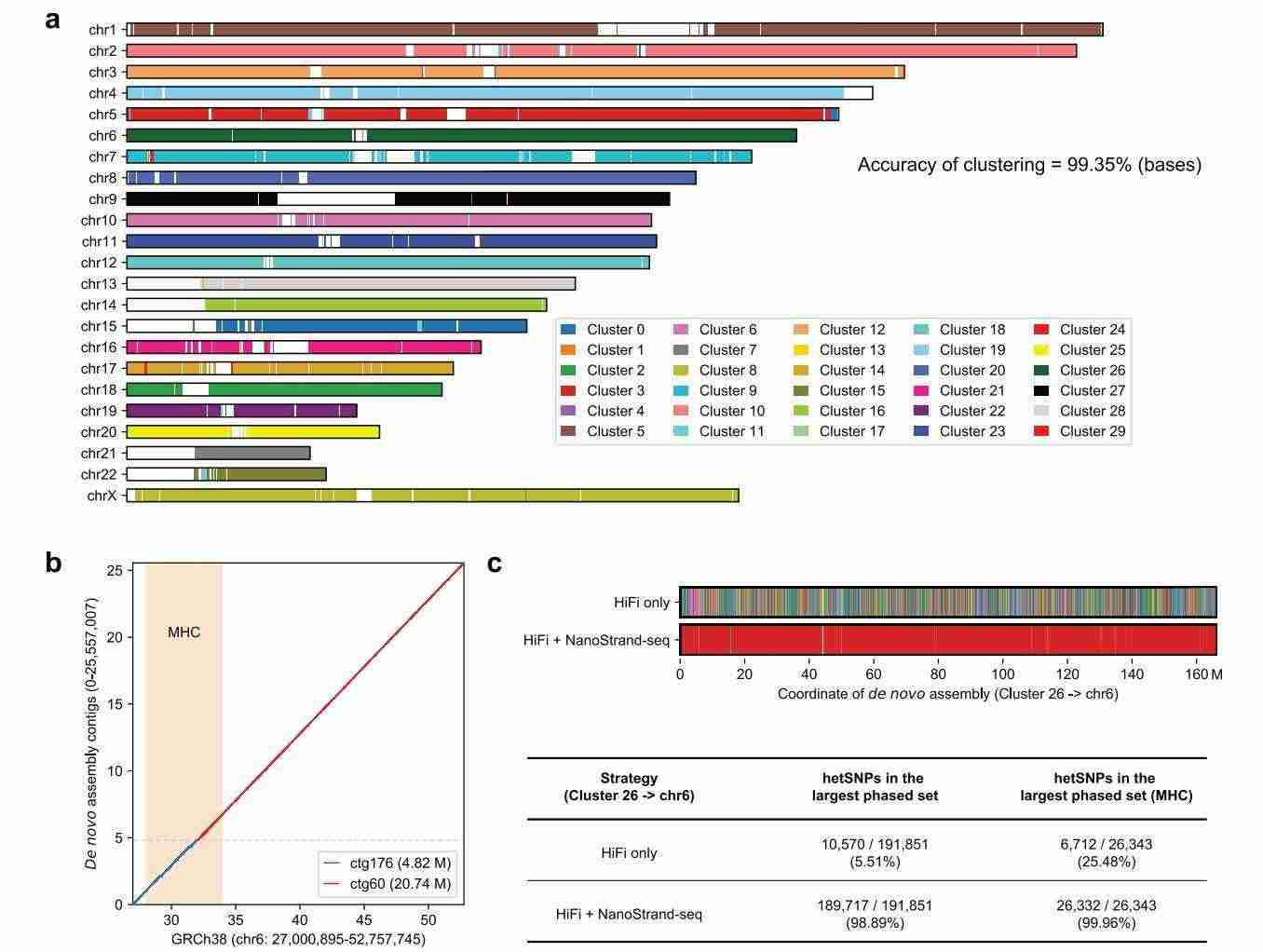 Haplotype-resolved de novo assembly by the combination of bulk HiFi long-read data and single-cell NanoStrand-seq data (Bai et al., 2024)
Haplotype-resolved de novo assembly by the combination of bulk HiFi long-read data and single-cell NanoStrand-seq data (Bai et al., 2024)
In the context of chromosome 6 of hg38, cluster 26 corresponds to a region of significant genetic diversity, with 98.89% of the heterozygous SNPs typed into the largest typing set. This set also includes the MHC region, which was assembled into two contiguous clusters. Notably, 99.96% of the heterozygous SNPs were typed into this largest typing set, underscoring the comprehensive coverage achieved. These results indicate that NanoStrand-seq can guide the de novo assembly of haplotyped genomes, especially complex genomic regions.
NanoStrand-seq enables precise haplotype typing of primary cells in short-term culture
In this study, NanoStrand-seq was successfully applied to short-term primary cultures of mouse embryonic fibroblasts and epithelial cells, and demonstrated excellent ab initio detection of SNPs and SVs at the whole chromosome level, as well as haplotyping.
 The performance of NanoStrand-seq with primary mouse cells (Bai et al., 2024)
The performance of NanoStrand-seq with primary mouse cells (Bai et al., 2024)
In conclusion, NanoStrand-seq has been demonstrated to possess the capability to accurately detect and ascertain the allelic profile of SNPs and SVs at the level of the entire chromosome in both primary cultures of a relatively short duration and in long-term passaged cultures. This method has been shown to be both highly accurate and reliable, particularly in the context of the analysis of complex genomic regions and the identification of structural variants. It is noteworthy that the method does not rely on neighbouring heterozygous SNPs for this purpose. Furthermore, NanoStrand-seq demonstrates a higher capacity for identifying known heterozygous SNPs, facilitating the assembly of the genome for haplotype typing from the beginning with a substantial amount of cellular HiFi data. The method is also capable of accurately assembling haplotypes in highly heterogeneous genomic regions.In addition to its capabilities, NanoStrand-seq possesses the potential to typify triploid, tetraploid and other polyploid genomes.
Reference
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.


