Insects play a vital role in ecosystem and agricultural production. As pollinators, decomposers and key links in the food chain, they maintain ecological balance and the stability of biodiversity. At the same time, many insects are the main crop pests, which bring great challenges to agricultural production. By deeply analyzing the structure and function of insect genome, we can better understand its physiology and behavior, reveal its gene function, gene regulation mechanism and adaptive evolution process, so as to develop more effective pest control strategies, promote the sustainable development of agriculture and ecosystem protection, promote the scientific progress of agriculture and ecosystem management, and finally provide scientific biological theoretical guidance for solving global food security and ecological protection problems.
Research Background
Pests have caused huge economic losses to crop production. With the rapid development of sequencing technology, especially the third generation of long reading and long sequencing technology has significantly improved the effect of genome assembly, the importance of genome sequencing and de novo assembly in promoting pest control and genomics research is increasingly apparent. Through genome analysis, scientists can deeply understand the genetics, adaptability, invasiveness and location of resistance genes of pest populations. However, high-quality genome assembly of non-model organisms still faces the challenge of high cost and complexity. In order to overcome these challenges, researchers deeply evaluated the assembly performance of different software on high-depth three-generation sequencing data sets, aiming at providing more reliable guidance and tool selection basis for future pest genome assembly, especially for LEPIDOPTERA projects.
Research Objects Species
Three different silkworm species: D9L, P50T, D9L X N4
PacBio CLR data, HiFi data, ONT data, Hi-C data
CLR data: Canu, NextDenovo, MECAT and wtdbg2
HiFi data: HiCanu, hifiasm
ONT data: NextDenovo, NECAT, wtdbg2
Service you may interested in
Research Conclusion
- Summary of original data and sequencing methods
In order to compare the performance of different TGS platforms in constructing highly continuous genome assembly of Lepidoptera pests. The researchers sequenced and analyzed the long reading data of three silkworm strains: (1)D9L strain, read by PacBio CLR, with 48 Gb data (110× coverage, N50 is 11,722 BP); (2)P50T strain, read by PacBio HiFi, with 27 Gb data (60× coverage, N50 is 15,818 BP); (3)D9L × N4 strain, read by ONT, 70 Gb data (160× coverage, N50 is 32,103 bp). Based on this, the researchers assembled the genome sequencing data with seven different assembly tools, and evaluated the assembly effect based on contig, Contig N50 length (N50), BUSCO Complete genes, quality value (QV) and other indicators.
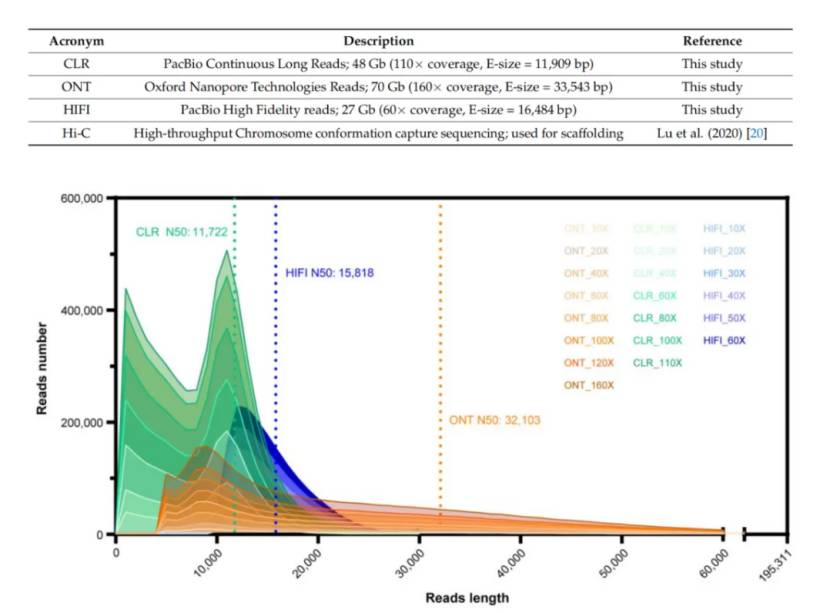 Sequence datasets and De novo assembly subsets with different data depths (Zhang et al., 2023)
Sequence datasets and De novo assembly subsets with different data depths (Zhang et al., 2023)
- ONT data genome assembly results
Researchers selected silkworm strain D9L × N4 with high genomic heterogeneity (about 1.11% heterozygosity) for ONT sequencing and assembly test. Three different long reading and long sequence assembly tools (NextDenovo, wtdbg2 and NECAT) and a subset of eight different coverage depths (10×, 20×, 40×, 60×, 80×, 100×, 120× and 160×) are used for assembly. The results showed that the genome assembled by NextDenovo was the smallest (about 449-468 Mb), the number of contig was about 89-114, and contig N50 was about 10.0-13.8 Mb, and it had the most complete BUSCO genes. The genome assembled by wtdbg2 is the largest, with a large number of contig, and the whole genome is fragmented. Although the assembly quality of wtdbg2 is not ideal, it is the only software that can be assembled at 10x sequencing depth, and it is the fastest software. The assembly quality of NECAT is between NextDenovo and wtdbg2, and the overall performance is moderate. The accuracy evaluation shows that NextDenovo has the least number of small-scale errors, and the number of structural errors is second only to wtdbg2. Wtdbg2 has the largest number of small-scale errors, but the least number of structural errors. NECAT has the largest number of structural errors and the second largest number of small-scale errors.
In addition, in order to study the influence of sequencing depth on assembly tools, the researchers evaluated the quality of ONT assembly at different sequencing depths (10×, 20×, 40×, 60×, 80×, 100×, 120× and 160×). The assembly quality of low-depth subsets (10× or 20×) varies greatly among different assembly tools. The 40× ONT dataset can complete most of the genome assembly, but deeper sequencing depth is needed to improve the genome quality.
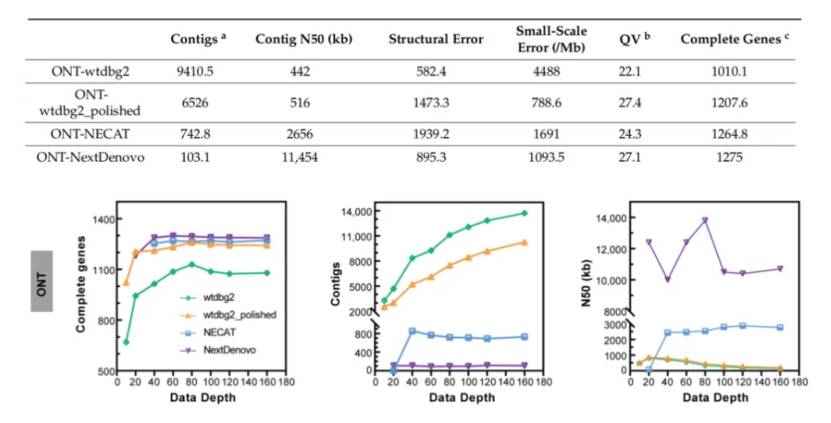 Comparison of ONT data assembly effect (Zhang et al., 2023)
Comparison of ONT data assembly effect (Zhang et al., 2023)
From the assembly results and evaluation results as a whole, NextDenovo software and 80x sequencing depth are ideal assembly options for ONT data.
- CLR data genome assembly results
The researchers selected silkworm D9L samples for testing. Four different long reading and long sequence assembly tools (NextDenovo, Canu, wtdbg2 and MECAT2) are used to assemble CLR readings. The results showed that when the sequencing depth reached 40× or higher, there was little difference in the number of contig assembled by each genome, and the result of NextDenovo was the best. Contig N50 increased with the increase of sequencing depth, and the increase of NextDenovo was the most significant. Wtdbg2 generates the least structural errors, followed by NextDenovo. NextDenovo generates the least small-scale errors, followed by Canu. NextDenovo has the highest continuity (contig N50 = 9.41 Mb), the smallest size (477 Mb), the least number of contigs (n = 205) and the largest assembly of Canu (506 Mb), but it has a high degree of repetition (2.9% of repeated BUSCOs). In this part of the study, the researchers found that about 40× CLR data set can construct most genomes, and increasing the sequencing depth can improve the quality of genomes, and whether polishing is needed depends on the assembly tools used.
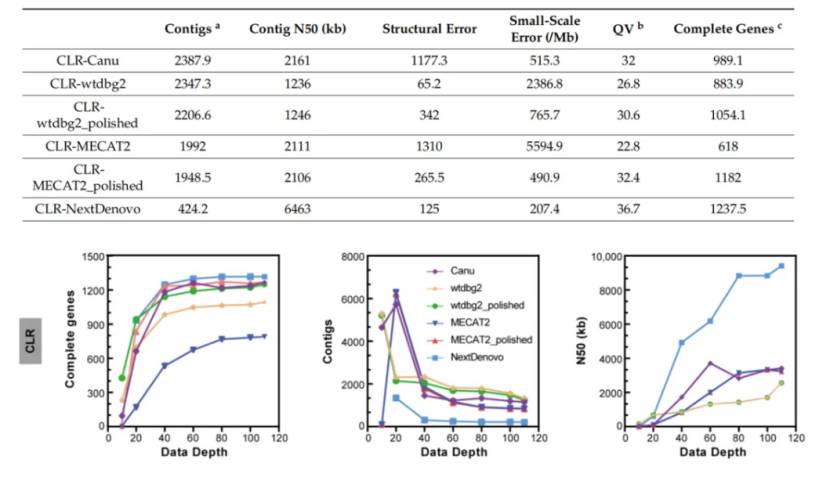 Comparison of CLR data assembly effect (Zhang et al., 2023)
Comparison of CLR data assembly effect (Zhang et al., 2023)
- HiFi data genome assembly results
The researchers selected P50T silkworm for PacBio HiFi sequencing, and used HiCanu and hifiasm for assembly. Compared with CLR and ONT assembly, HiFi assembly has superior genomic continuity and integrity. There is no significant difference in the size, continuity and completeness of HiFi assembly between the two softwares, but the biggest difference lies in the number of contigs, in which the number of contigs assembled by hifiasm is much smaller than that assembled by HiCanu. Compared with ONT and CLR assembly, HiFi assembly contains the least structural errors and small-scale errors. In addition, the researchers evaluated the assembly quality of HiFi at different sequencing depths (10×, 20×, 30×, 40×, 50× and 60×) to study the influence of data depth on assembly tools. It is found that about 20× HiFi data set can generate most genomes. Because HiFi only needs 20× or higher sequencing depth to construct most of the genome, and no subsequent polishing process is needed, the time required for genome assembly is much less than ONT and CLR.
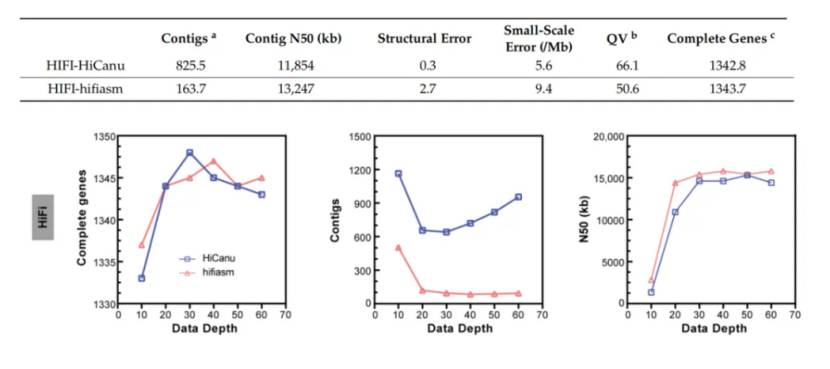 Comparison of HiFi data assembly effect (Zhang et al., 2023)
Comparison of HiFi data assembly effect (Zhang et al., 2023)
Research Summary
PacBio HiFi data is obviously superior to CLR and ONT in the continuity and integrity of the genome assembled based on HiFiasm software. And less memory and machine resources are consumed, so that the assembly process is more convenient, and no additional polishing process is needed. From the sequencing depth, 30x HiFi data can reach a relatively stable and high-quality Contig N50 level. To sum up, based on the data in this paper, HiFi ASM assembly based on 30x HIFI data may be the best choice for genome sequencing of Lepidoptera pests in the future. In addition, for other types of insect genome assembly, the conclusion of this paper also has high reference value. Insect products The third-generation DNA library sequencing products and genome De novo products have been deeply cultivated in the industry for many years. The third-generation HiFi library sequencing cycle only takes 7 days, the single Cell output of insect HiFi data can exceed 94 G, and the average yield of the project exceeds 100%.
The three-generation library sequencing of butterflies, moths, moths, bees and other insects has been completed, with rich experience and stable data quality. Insect micro-library-building sequencing products support a minimum of 5ng DNA investment, and a single insect can complete extraction, library building and HiFi sequencing, which is the best choice for your small and micro-insect assembly.
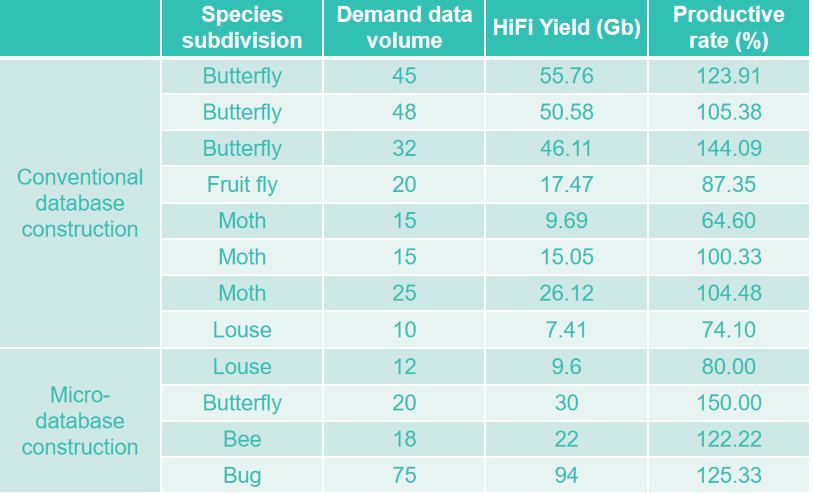 PacBio HiFi sequencing output of insect samples
PacBio HiFi sequencing output of insect samples
Reference
-
Zhang Tong, Xing Weiqing, Wang Aoming, Zhang Na, Jia Ling, Ma Sanyuan and Xia Qingyou. "Comparison of Long-Read Methods for Sequencing and Assembly of Lepidopteran Pest Genomes." Int. J. Mol. Sci. (2023) 24: 649. https://doi.org/10.3390/ijms24010649
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.






