

We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
DNA affinity purification sequencing (DAP-seq) represents an efficient in vitro approach for identifying transcription factor binding sites (TFBS). In this technique, the transcription factor (TF) expressed exogenously and genomic DNA are purified. Subsequently, the DNA fragments bound by the TF are subjected to high-throughput sequencing, and the TFBS within the specific DNA fragments are determined through the analysis of the resulting sequence data.
DAP-seq technology capitalizes on tag proteins, such as TFs, expressed in vitro. By incubating purified proteins with target genomic DNA fragments, the protein-binding sequence fragments can be ascertained. This method obviates the need for specific antibodies and transgenic manipulation, thereby enabling its application in species lacking a stable genetic transformation system and for target proteins without specific antibodies. However, it is important to note that DAP-seq is an in vitro experiment, and the genomic DNA with binding fragments lacks the spatial and structural information present in vivo. Consequently, it may not accurately reflect the in vivo interaction between transcription factors and DNA.
Service you may interested in
Construction of genome library: High-quality genomic DNA is initially extracted from samples and then subjected to physical or enzymatic digestion to generate DNA fragments. Subsequently, a genomic DNA library is constructed by ligating sequencing adapters based on the Illumina platform. This step is crucial for maintaining the integrity of the DNA fragments and lays the foundation for subsequent high-throughput sequencing.
In vitro expression and purification of TF: The coding DNA sequence (CDS) encoding the target TF is cloned into an expression vector harboring an affinity tag, such as Halo Tag. Following construction, the vector is introduced into an appropriate expression system for in vitro protein expression. This enables the production of TF proteins fused with affinity tags, which are then available for subsequent affinity purification and analysis.
Affinity purification and sequencing: After purifying the expressed TF-affinity tag fusion protein, it is incubated with the pre-constructed genomic DNA library. The TF within the fusion protein can specifically bind to the target sequence on the DNA fragment. Affinity purification is then carried out using Halo Tag-specific magnetic beads, and the protein-DNA complex bound to the target DNA is isolated from the mixture. The captured DNA fragments are amplified by PCR and finally sequenced using the Illumina high-throughput sequencing platform to obtain high-quality sequence data.
Data analysis: The obtained sequencing reads are aligned with the reference genome to identify the potential binding sites of the target transcription factors. Through bioinformatics analysis, the distribution of these binding sites within the genome and their potential functions in regulating gene expression can be further elucidated. This aids in a more profound understanding of the biological function and regulatory mechanism of the TF.
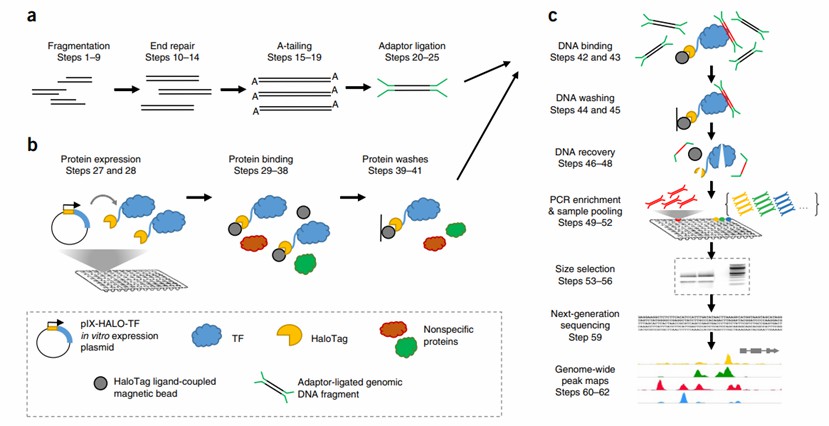 Overview of DAP-seq process (Anna et al., 2017)
Overview of DAP-seq process (Anna et al., 2017)
In the realm of genomics and epigenetics research, the discovery of TFBS has been a focal point. The traditional Chromatin Immunoprecipitation sequencing (ChIP-seq) method can effectively detect TFBS when high-quality antibodies are available. However, the scarcity of such antibodies has limited the broader application of ChIP-seq. The emergence of DAP-seq technology has liberated TFBS research from the constraints of species and antibody quality, providing a novel and powerful tool for TF research in the life sciences.
In a study published on October 11th, 2022, by a research group from the Synthetic Biology Research Center of the Shenzhen Institute of Agricultural Genome, Chinese Academy of Agricultural Sciences, in The Plant Journal, the allelic genes of tea hybrids were identified and analyzed for bias. Through the combined use of ATAC-seq and DAP-seq, the authors unraveled the regulatory mechanisms of allelic genes and chromatin in tea hybrids, offering novel insights into the formation and regulation of tea heterosis.
Research sample
Tea plants (Parent: Tie Guanyin, Huang Dan; Hybrid: Jin Guanyin, Huang Guanyin).
Research methods
Service you may interested in
Results and conslusions
To investigate the regulation of allelic parental bias and chromatin accessibility in tea heterosis, the authors analyzed the whole-genome RNA-seq and ATAC-seq of several tea plants. The results indicated that the parents had a significant impact on the allelic gene expression in the tea hybrids. To further explore the parental bias in the tea hybrids, the authors systematically identified the ASEGs in the hybrids. In the buds of the hybrid Jin Guanyin, a total of 4827 ASEGs were identified, with 2250 from the paternal Tie Guanyin and 2577 from the paternal Huang Dan. Similar parental bias was also observed in Huang Guanyin. These findings suggest that the majority of ASEGs exhibit conserved parental bias in expression. Additionally, the authors performed KEGG enrichment analysis on the ASEGs with different parental biases in the hybrids. The results demonstrated that parental bias had a substantial influence on the growth and metabolism of the tea hybrids.
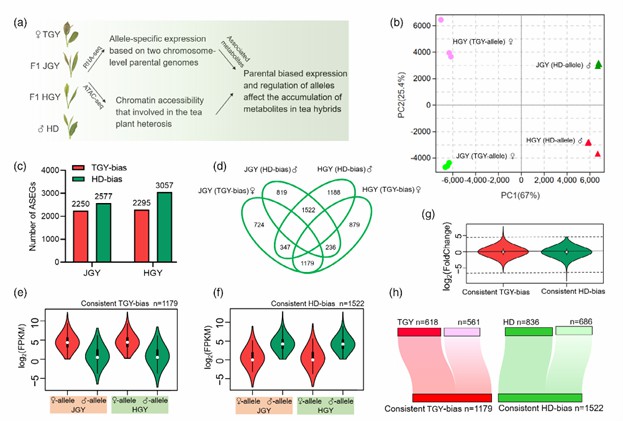 Global analysis of alleles and ASEGs with parental bias in tea plant hybrids (Wang et al., 2022)
Global analysis of alleles and ASEGs with parental bias in tea plant hybrids (Wang et al., 2022)
To study the regulation of chromatin accessibility on ASEGs with positive heterosis, the authors identified 8 and 16 ACRs in 17 and 15 ASEGs with terpenoid and purine alkaloid metabolic pathways, respectively. The frequency and accessibility of ASEGs in purine and purine alkaloid metabolic pathways were significantly higher than those of ACRs in terpenoid metabolic pathways. In the purine nucleotide and alkaloid metabolic pathways, the average abundance of ACRs in the hybrid Huang Guanyin was higher than that in other tea varieties.
In addition, the authors selected three ACRs with high accessibility in the terpene synthesis pathway of the tea hybrids for visualization research on the IGV browser. The results showed that these three ACRs contained multiple potential binding sites of MYB, ARF, and NAC. It has been reported that many homologous TF members of these targets are involved in the activation of numerous structural genes in the terpenoid synthesis pathway and affect the terpenoid content. To study these targets, the binding site of CsMYB83 was identified using DAP-seq technology. The results indicated that CsMYB83 had a binding peak at the ACRs of CsDXS, suggesting that CsMYB83 might have a heterosis regulatory role at the ACRs.
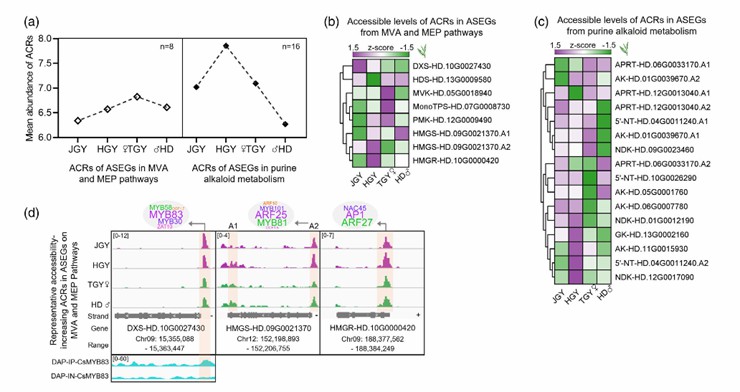 Analysis of ACRs in ASEGs (Wang et al., 2022)
Analysis of ACRs in ASEGs (Wang et al., 2022)
A research paper titled "Deciphering the regulatory network of the NAC transcription factor FvRIF, a key regulator of strawberry (Fragaria vesca) fruit tearing" was published in The Plant Cell in 2023. The authors discovered that FvRIF in the diploid strawberry Fragaria vesca is a crucial regulator of fruit ripening, and the knockout mutation of FvRIF leads to the failure of fruit maturation and development. The combination of DAP-seq and transcriptome analysis revealed that 2080 genes are the direct targets of FvRIF-mediated regulation, encompassing genes related to all aspects of fruit ripening.
Research samples
Diploid strawberry (Fragaria vesca cv. Ruegen) and octoploid strawberry (Fragaria × Ananassaduch cv. Beni Hoppe).
Research methods
Service you may interested in
Results and conclusions
To gain a better understanding of the transcriptional regulation of fruit ripening mediated by FvRIF, the authors conducted DAP-seq to identify the binding sites of FvRIF. A total of 9902 enrichment peaks were detected in two biological replicates, corresponding to 7899 genes, and the binding sites were highly concentrated around the transcription initiation site (TSS). The majority of the binding sites (37.0%) were located in the promoter region (2kb upstream of TSS), consistent with the fact that FvRIF is a TF with DNA binding and gene regulatory activities.
Four enriched motifs were identified using the Homer tool. The most abundant motif was represented by TACGTACGTAAC, accounting for 4649 FvRIF binding peaks. The sequence of this motif is CGT[G/A], which is a previously reported NAC recognition motif. This result validates the reliability of the DAP-seq data. The authors also identified three other motifs, TAGCTA(A/G)C, CTTC(A/G)TTT, and AGAAAGAA, which were present in 4,473, 3,500, and 2,222 FvRIF binding peaks, respectively. The GO enrichment analysis of the genes containing FvRIF binding sites indicated that they are primarily involved in transcriptional regulation, plant hormone-mediated signaling pathways, cell wall tissue or biogenesis regulation, and development and maturation processes. This suggests that FvRIF may directly control various biological pathways related to fruit ripening.
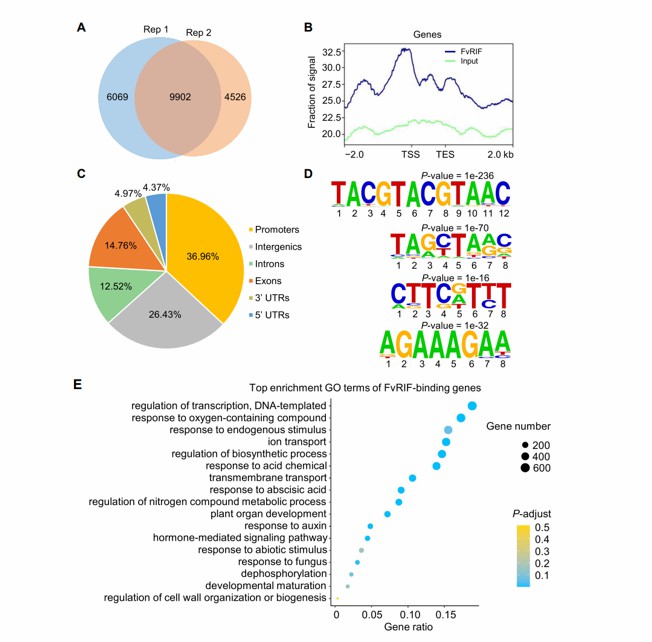 Genome-wide identification of FvRIF binding sites through DAP-seq (Li et al., 2023)
Genome-wide identification of FvRIF binding sites through DAP-seq (Li et al., 2023)
By identifying the binding sites of TF in the whole genome, we can more comprehensively understand how TF choose specific sites on the genome to bind, so as to achieve fine regulation of gene expression, which is of great significance to reveal the mechanism of TF in various biological processes. DAP-seq can identify TFBS efficiently and accurately by combining DNA- protein interaction experiment with high-throughput sequencing. DAP-seq can reconstruct DNA- protein interaction in vitro, which makes the experiment easy to operate and can eliminate the interference of cell environment on binding events.
References
 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.
CD Genomics is propelling the future of agriculture by employing cutting-edge sequencing and genotyping technologies to predict and enhance multiple complex polygenic traits within breeding populations.


