
 Sample Submission Guidelines
Sample Submission Guidelines
CD genomics provides a ribosome-profiling strategy that is based on the deep sequencing of ribosome-protected mRNA fragments and enables genome-wide investigation of translation with sub cordon resolution. Using nuclease digestion, the ribosome position and the translated message can be precisely determined by analyzing the protected ~30-nt area of the mRNA template.
What is Ribo-seq
Ribo-seq, or "Ribosome profiling," refers to the sequencing of actively translating RNA fragments that are associated with ribosomes. This technique provides precise and quantitative information about all translatable molecules in a sample, including mRNAs and other potentially translatable RNA molecules such as lncRNAs and circRNAs.
Ribo-seq is one of the most commonly used methods in translatome sequencing. This technique involves digesting cellular RNA using RNases to obtain ribosome-protected RNA fragments that are actively being translated, often referred to as "ribosome footprints" (RFs). These RFs are typically around 30 nucleotides in length. Subsequently, these ribosome-protected fragments are enriched, deeply sequenced, and analyzed. Ribo-seq provides information at the whole-genome level about protein translation efficiency, enables the discovery of new proteins or short peptides, and allows researchers to calculate the translation efficiency of RNA in conjunction with transcriptome data. By combining this data with proteomics information, the relationship between transient translation and protein accumulation can be analyzed, aiding in the study of protein degradation effects. Translatome sequencing serves as a bridge between RNA and protein, enabling the investigation of various aspects of gene translation, such as levels, regions, and rates. When combined with transcriptome data, small RNA sequencing, proteomics, and other analyses, it facilitates more precise research into post-transcriptional and translational regulation mechanisms.
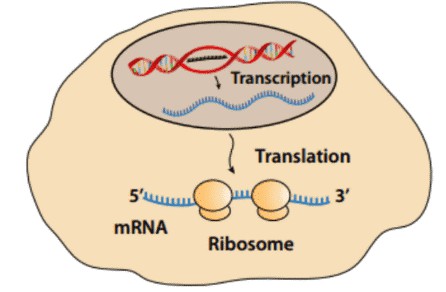 Fig.1 Ribosome-protected mRNA
Fig.1 Ribosome-protected mRNA
Advantages of Our Ribosome Profiling Service
- By identifying actively translating transcripts, Ribo-Seq can provide visibility into important aspects of gene regulation.
- Study translation control and measure gene expression.
- Identify translation initiation sites.
- Determine protein synthesis rates.
- Predict protein abundance.
- Fast turnover, high quality, and high stability.
- Professionally designed protocols with strict quality control.
Application of Ribosome Profiling
- Sequences of ribosome-protected mRNA.
- The study of active mRNA translation with sequencing.
- Prediction of protein abundance.
- Identify translation start sites
- Investigation of transcriptional control and post-transcriptional regulation.
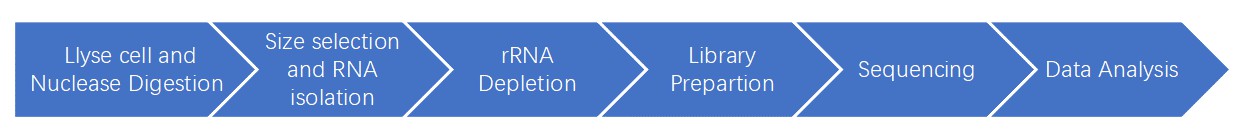
Ribosome Profiling Workflow
Using nuclease digestion to isolate nuclease-resistant ribosome, the translated message can be precisely determined by analyzing the protected 30 nucleotides during translation.
Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
|
Ribo-seq Research Strategy
Deliverables
- Experimental procedure
- Experiment parameter
- Original data
- Results of bioinformatic analysis
References:
- Ingolia N T, Hussmann J A, Weissman J S. Ribosome profiling: global views of translation. Cold Spring Harbor perspectives in biology, 2019, 11(5): a032698.
- Olexiouk V, Crappé J, Verbruggen S, et al. sORFs. org: a repository of small ORFs identified by ribosome profiling. Nucleic acids research, 2016, 44(D1): D324-D329.
- McGlincy N J, Ingolia N T. Transcriptome-wide measurement of translation by ribosome profiling. Methods, 2017, 126: 112-129.
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
1. What is ORF?
The critical aspect of Ribo-seq, akin to RNA-seq, lies in the identification of Open Reading Frames (ORFs), enabling the recognition of mRNA, lncRNA, and circRNA. An ORF represents a series of codons starting with the initiation codon, typically AUG, and terminating at the stop codon, which is traditionally UAA, UAG, or UGA.
2. What is the protocol for ribosome profiling?
- Harvest cells or tissues and treat them with a translation inhibitor to freeze ribosomes on mRNA molecules at different positions.
- Lyse cells to release ribosomes and mRNA, followed by nuclease treatment to digest unprotected RNA.
- Separate ribosome-protected mRNA fragments (RPFs) using sucrose gradient centrifugation or size selection.
- Construct a cDNA library from RPFs, typically through reverse transcription and PCR amplification.
- Sequence the cDNA library using high-throughput sequencing technologies to determine the positions of ribosomes on mRNA transcripts.
- Analyze sequencing data to map ribosome footprints to the transcriptome, identify translation initiation sites, calculate translation rates, and study translational dynamics and regulation.
3. What are the limitations of ribosome profiling?
- The experimental rRNA residue is high
- Ribosome footprints are short, making it difficult to identify genuine ORFs
- Necessitates inference of protein synthesis rates
- The main limitation of ribosome profiling analysis is the requirement for relatively large amounts of samples
4. What is the difference between RNA-Seq and ribosome profiling?
RNA sequencing is a technique widely used for transcriptomic research, capable of determining the types and relative abundance of all RNAs present in a cell or tissue. This process measures the total quantity of RNA molecules, encompassing mRNAs, non-coding RNAs, and other RNA varieties. Meanwhile, ribosome profiling is a technique employed for investigating the translatome, focusing on the location and actions of ribosomes during protein synthesis. It identifies mRNAs undergoing translation by capturing mRNA fragments protected by ribosomes and unveils the positions of ribosomes on the transcript. In summary, while RNA sequencing is applicable for a comprehensive understanding of overall gene expression, ribosome profiling places an emphasis on detailing the process and regulatory mechanisms of protein synthesis.
The Translational Landscape of the Human Heart
Journal: Cell
Impact factor: 36.216
Published: 2019 May 30 2019
Backgrounds
Gene expression in human tissues has predominantly been studied at the transcriptional level, largely overlooking regulatory elements at the translation level. In this context, the authors have analyzed the translatome of 80 human hearts with the intent to identify novel translational events and quantify the impact of translational regulation.
Methods
- TruSeq Ribo Profile Library Prep Kit
- Illumina HiSeq 2500
- Total RNA sequencing
- mRNA sequencing
- Gene and ORF quantification
- Ribosome-associated circRNAs
- Quantification analyses
- Statistical analyses
Results
1. A Snapshot of Active Translation in 80 Human Hearts
In an endeavor to explore the expression and translation of mRNA in cardiac tissues, the authors conducted mRNA sequencing (mRNA-seq) and ribosome sequencing (Ribo-seq) on left ventricular tissue from 65 end-stage DCM patients and 15 non-DCM controls. The inferred quantity of translated gene products from the authors' ribosomal sequencing data was twice that of mRNA sequencing (Figure 1F), possibly owing to the occlusion of mass spectrometry detection of lowly expressed proteins by highly expressed cardiac myofilament proteins. The predictive value of Ribo-seq for final protein levels exceeds that of mRNA-seq.
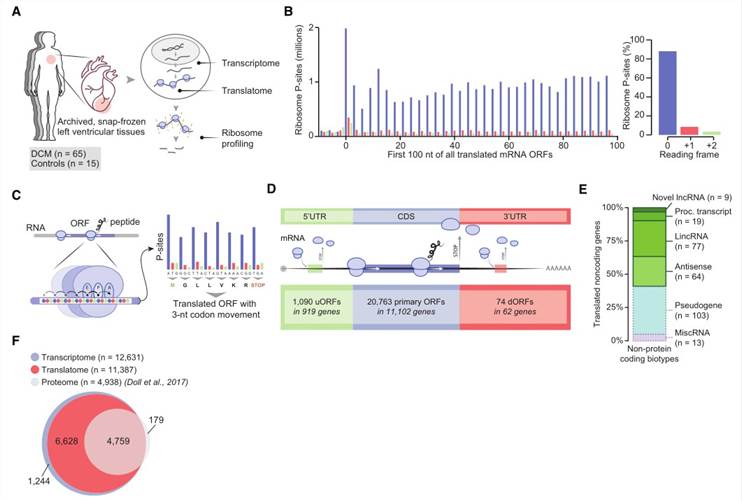 Figure 1. A Snapshot of Active Translation in 80 Human Hearts
Figure 1. A Snapshot of Active Translation in 80 Human Hearts
2. Upstream ORFs Influence Translational Efficiency Independent of Translation Rates
The crux of this study was the detection of actively translated upstream open reading frames (uORFs) in the heart tissue transcriptome. The authors identified 1,090 actively translated uORFs spanning across 919 genes, accounting for 8% of all translated genes. Against expectations, there was an overall mild positive correlation observed between the translation rates of these uORFs and the primary ORFs, rather than a linear inverse relationship suggesting reduction in translation efficiency. The inference drawn was that for the majority of these uORFs, no quantitative dependency could be detected between the uORF translation frequency and the observed decrease in the primary ORF translation efficiency.
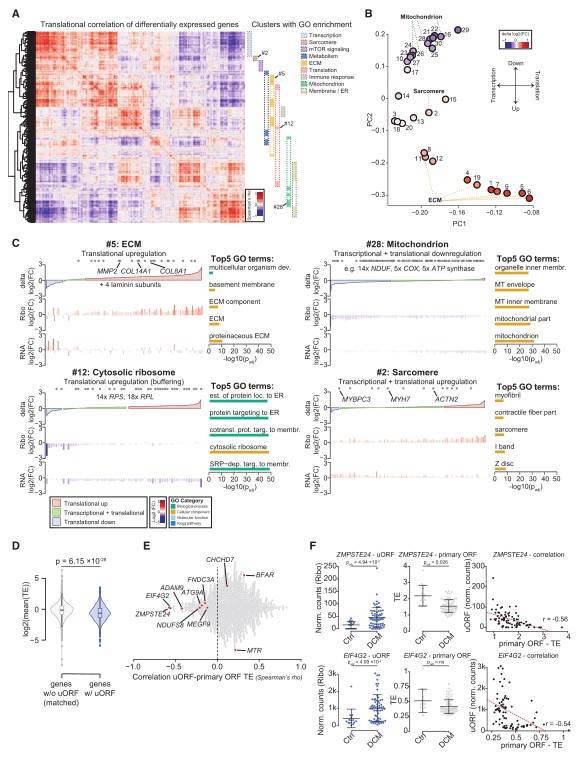 Figure 2. Dissecting Transcriptional and Translational Control in Human Tissue
Figure 2. Dissecting Transcriptional and Translational Control in Human Tissue
3. Expression Regulation of Translated lncRNAs in Healthy and Diseased Hearts
The authors noted a general reduction in co-regulation during the translation process for most gene pairs, with the exception of TRDN-TRDN-AS1. Among all translated lncRNAs, 34 were found to be upregulated and 7 downregulated in diseased hearts, highlighting a need for further exploration into the potential roles of these microproteins.
4. Translation of Human Cardiac circRNAs
In their research, the authors identified 8,878 human cardiac circular RNAs (circRNA) in 3,181 genes, 2,070 of which had never been detected previously. Furthermore, ribosome association revealed potential protein translation from 40 circRNAs produced by 39 genes (Figure 3A). Notably, newly detected ribosome-associated circRNAs included the heavily occupied circCFLAR (Figure 3D) and the heart-specific circRNAs such as circSLC8A1 (Figure 3E), circMYBPC3, and circRYR2. Additionally, one of the ribosome-associated circRNAs was the sponge circRNA, circCDR1-AS.
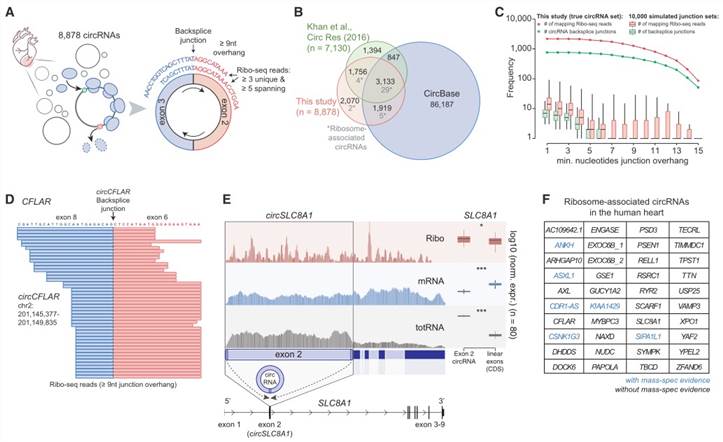 Figure 3. Translation of Human Cardiac circRNAs
Figure 3. Translation of Human Cardiac circRNAs
Conclusion
Remarkably, a wide array of microproteins originates from the translation of long non-coding RNAs (lncRNA), which exhibit salient traits in both health and disease contexts. Our research suggests that numerous translated lncRNAs may have a dual function, possessing both coding and non-coding roles. This sheds new light on the complexity and versatility of cellular regulation mechanisms. Ribosome profiling reveals the principles of translational control in human tissue. Ribosomes translate mRNAs downstream of protein-truncating variants. Functionally characterized lncRNAs and circRNAs produce microproteins in vivo.
Reference:
- van Heesch S, Witte F, Schneider-Lunitz V, et al. The translational landscape of the human heart. Cell, 2019, 178(1): 242-260. e29.
Here are some publications that have been successfully published using our services or other related services:
Disruption of tRNA biogenesis enhances proteostatic resilience, improves later-life health, and promotes longevity
Journal: Plos Biology
Year: 2024
Circular DNA tumor viruses make circular RNAs
Journal: Proceedings of the National Academy of Sciences
Year: 2018
Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal: Journal of Autoimmunity
Year: 2024
Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Journal: Neuropsychopharmacology
Year: 2024
FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Journal: Elife
Year: 2023
Identification of circular RNAs regulating cardiomyocyte proliferation in neonatal pig hearts
Journal: JCI insight
Year: 2024
See more articles published by our clients.


