We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Fiber-seq Service
Chromatin structure plays a crucial role in gene expression, genome stability, and cellular function. However, traditional chromatin analysis methods rely on fragmented DNA, making it challenging to accurately pair DNA sequences with chromatin accessibility on single molecules. With the advent of third-generation sequencing, long-read platforms like PacBio have revolutionized the study of genome organization and epigenetics. Single-Molecule Chromatin Fiber Sequencing(Fiber-seq) enables direct detection of chromatin accessibility at single-molecule resolution, providing a deeper understanding of non-coding regulatory elements and complex genomic structures. Utilizing this advanced technology, CD Genomics offers Fiber-seq, delivering high-resolution insights into chromatin dynamics to support epigenetic and genomic research.
What is Fiber-seq
In 2020, John A. Stamatoyannopoulos from the Altius Institute for Biomedical Sciences and Andrew B. Stergachis from Harvard Medical School collaborated to introduce the Fiber-seq technology in Science. Fiber-seq enables the study of regulatory DNA and nucleosome positioning at single chromatin fiber resolution by utilizing m6A methyltransferase (MTase) to label open chromatin regions, combined with PacBio HiFi long-read sequencing.
The experimental workflow of Fiber-seq includes nucleus extraction, MTase reaction, purification, DNA fragmentation, library construction, and sequencing. During data analysis, the methylation-accessible sites (m6A sites) detected through base chemical modifications allow the identification of nucleosome positioning and open chromatin. This information can be further used to analyze transcription factor footprints, co-activation states of different genomic regions, and precise haplotype chromatin structures.
Moreover, a single Fiber-seq experiment can simultaneously capture genomic sequences, DNA methylation patterns, and chromatin accessibility, enabling a comprehensive analysis of the gene regulatory landscape.
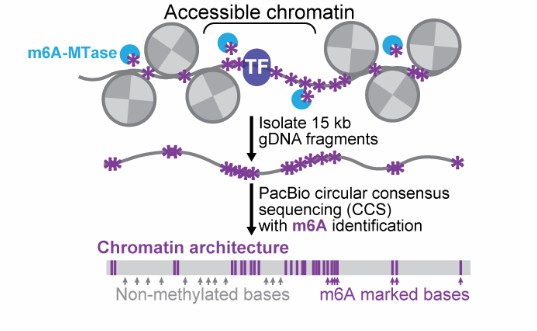 Figure 1. Principle of Fiber-seq technology.(Stamatoyannopoulos et al. 2020)
Figure 1. Principle of Fiber-seq technology.(Stamatoyannopoulos et al. 2020)
Advantages of Fiber-seq
More Accurate Detection
Compared to other chromatin accessibility detection techniques, Fiber-seq does not require enzymatic digestion. Instead, it directly labels open DNA regions, enabling the analysis of DNA sequences on individual chromatin fibers and the identification of accessible chromatin regions. This approach effectively reduces false positives, leading to more accurate detection.
Haplotype-Level Analysis of Gene Regulatory Mechanisms
Leveraging long-read sequencing technology, Fiber-seq can simultaneously reveal chromatin structural and functional differences between haplotypes, offering a novel perspective on the complexity of gene regulation.
Deciphering Chromatin Structures in Complex Regions
Third-generation long-read sequencing can cover complex genomic regions, such as telomeres and highly repetitive sequences, allowing for a detailed analysis of chromatin structure and uncovering how regulatory elements coordinate transcriptional activities in these regions.
Unlocking Multi-Omics Data
A single library enables access to multiple omics datasets, preventing batch effects caused by the independent preparation of different omics libraries.
Fiber-seq Sample Requirements
Sample Type: Fresh or flash-frozen cells, or nuclei isolated from cells/tissues, ensuring high molecular weight DNA (if applicable).
Cell Quantity and Viability: A minimum of 1 million cells is required, with an optimal range of 5–10 million cells, and cell viability should be ≥ 90%.
Storage and Transportation: Cells should be stored at -80°C or in liquid nitrogen, while nuclei can be stored in a storage buffer at -80°C.
Quality Control: Ensure the sample is free of contamination and aggregation, with intact DNA and no RNase contamination.
Workflow of Our Service
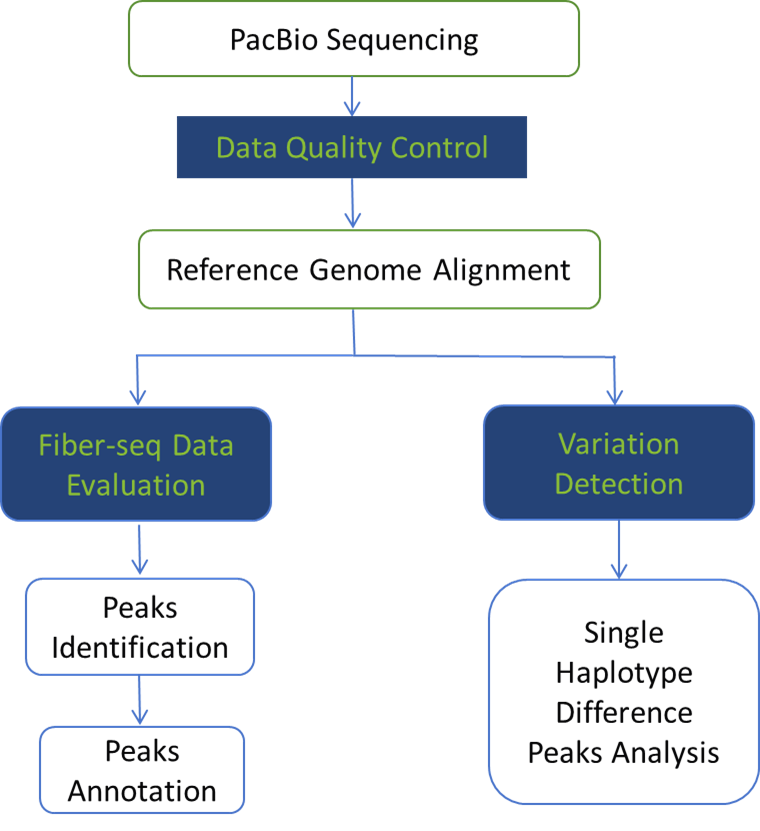
Analysis Contents
- Data Quality Control
- m6A Site Prediction and Statistics
- MSP Prediction and Statistics
- Nucleosome Prediction and Statistics
- Sample Correlation Statistics
- Peaks Identification and Statistics
- Peaks Length Statistics
- Peaks Chromosome Distribution Statistics
- Peaks Accessibility Score Statistics
- Functional Region Distribution Statistics
- GO Enrichment Analysis
- ENCODE Database Analysis
- KEGG Enrichment Analysis
- Motif Analysis
- SNV/InDel Detection
- SNV/InDel Variation Detection Phasing
If you are interested in epigenetics research, gene regulation mechanisms, or analyzing complex genetic variations, the Fiber-seq technology will provide you with unprecedented precision. Using PacBio HiFi long-read sequencing technology, Fiber-seq demonstrates unique advantages in chromatin accessibility, complex genomic regions, and haplotype analysis, making it an indispensable tool for researchers.
Whether you're engaged in basic scientific research or disease studies, CD Genomics is here to provide you with professional support and customized services. If you are interested in Fiber-seq technology or have any questions, feel free to contact us anytime!
Case Study:Fiber-seq found that STR mutations activate tissue-specific enhancers
Background
Thyroid Stimulating Hormone (TSH) is the main regulator of thyroid gland growth and function. TSH resistance (RTSH) refers to a reduced sensitivity to TSH, characterized by elevated TSH levels but normal thyroid hormone levels. This condition may lead to hypothyroidism or thyroid enlargement. Hereditary RTSH has been found to be associated with a locus on chromosome 15q, but its genetic basis has not yet been clearly identified.
Methods
Whole Genome Sequencing (WGS): Whole genome sequencing was performed on two affected individuals from five RTSH families linked to the 15q locus to identify non-coding variants associated with RTSH.
Fiber-seq: Used to study chromatin accessibility in thyroid tissue and reveal how STR mutations affect gene regulation.
RNA Sequencing (RNA-seq): Analyzed gene expression differences in thyroid tissue to identify transcriptomic changes associated with STR mutations.
Sanger Sequencing: Used to confirm the STR variants identified by WGS and screen for STR variants in other RTSH families.
Results
Through Fiber-seq and RNA-seq, the study found that the mutated STR can activate a set of thyroid-specific enhancers, leading to haplotype-specific upregulation at the bidirectional MIR7-2/MIR1179 locus located 35 kb downstream, resulting in the overexpression of the corresponding microRNA products in thyroid cells. The signaling pathway imbalance targeted by these microRNAs provides a working model to explain the etiology of this form of RTSH.
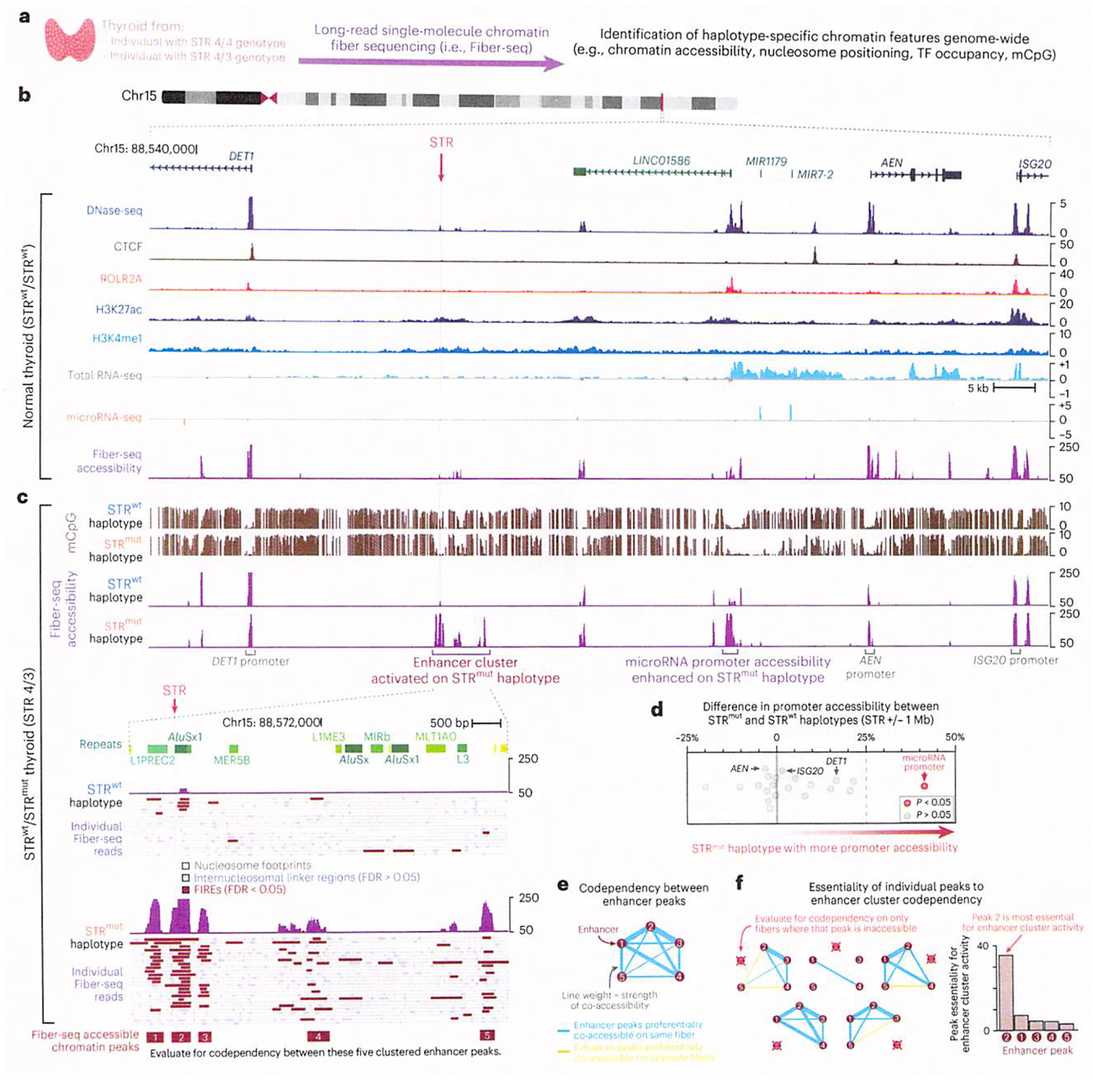 Figure 2. STRmut is associated with the activation of a codependent thyroid-specific enhancer cluster.(Grasberger et al. 2024)
Figure 2. STRmut is associated with the activation of a codependent thyroid-specific enhancer cluster.(Grasberger et al. 2024)
References
- Stamatoyannopoulos, J. A et al. (2020). Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science , 368(6498), 1449–1454. https://doi.org/10.1126/science.aaz1646
- Grasberger, H et al. (2024). STR mutations on chromosome 15q cause thyrotropin resistance by activating a primate-specific enhancer of MIR7-2/MIR1179. Nature genetics, 56(5), 877–888. https://doi.org/10.1038/s41588-024-01717-7
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment