

We are dedicated to providing outstanding customer service and being reachable at all times.

Animal/Plant Whole Genome Sequencing
CD Genomics' animal and plant whole genome sequencing, also known as animal and plant whole genome resequencing (WGRS), namely sequencing the entire genome of a plant or animal and aligning it to a known reference genome. We have two long-read sequencing platforms, PacBio SMRT sequencing and ONT nanopore sequencing, which provide comprehensive access to coding and non-coding regions of chromosomal, mitochondrial DNA, and chloroplast (plant) DNA.
Overview of Animal and Plant Whole Genome Sequencing

Animal and plant whole genome sequencing (WGS) is a powerful technique that is commonly used to achieve an accurate and comprehensive examination of the genomes of animals and plants. WGS methods offer many advantages over traditional methods such as genotyping sequencing and arrays, including the generation of orders of magnitude more data, relatively enhanced variant discovery, greater statistical power, and easier analysis. This technology can reveal the molecular mechanisms of breeding and species formation, identify common genetic variations among populations, and provide resources for accelerated genetic development. As a result, animal and plant whole genome sequencing has been used for a wide range of applications, including the study of the origin and evolution of species, genome-wide association studies (GWAS), and agricultural breeding projects.
Sample Requirements
- Genomic DNA amount:
- For additional sample inquiries, please contact our professional technicians.
-PacBio Sequel II sequencing platform: HMW Genomic DNA≥ 15μg, OD260/280 =1.8 ~2.0, A260/230=1.5 ~2.6
-Nanopore PromethION sequencing platform: HMW Genomic DNA≥ 8μg, OD260/280 =1.75 ~2.0, A260/230=1.4 ~2.6
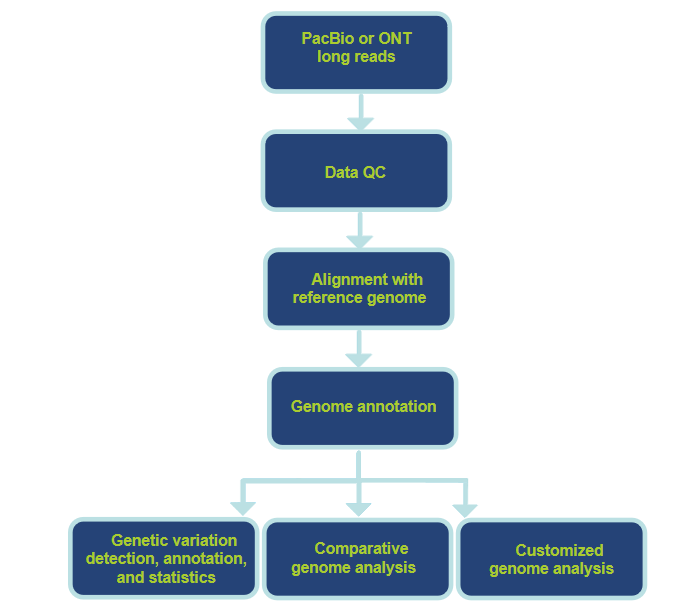
Workflow of Our Animal/Plant Whole Genome Sequencing Services
CD Genome provides professional services from experiment consultation and design, library construction and quality control (QC), long-read sequencing to data analysis. Each step is carefully designed and follows high scientific standards to ensure the reliability and accuracy of study results.

Analysis Pipeline and Contents
- Raw data processing.
- Standard analysis.
-Aligning to the current reference genome.
-Alignment statistics, including depth distribution and coverage calculation.
-Genetic variation detection, annotation, and statistics. - Advanced analysis.
-Comparative genome analysis.
-Customized genome analysis.
Benefits of Our Services
-Decades of experience in genomics services and innovative technology platforms.
-Flexible sequencing strategies and customized bioinformatics pipelines.
-Supporting a broad range of species.
-Fast turnaround time and complete analytical study reports.
-Most competitive prices.
We have years of experience in providing accurate and customized whole genome sequencing services and high-quality results. Based on advanced long-read sequencing platforms, our service generates reference-quality assemblies of complex plant and animal genomes by overcoming problems such as high GC regions that cannot be spanned, highly repetitive fragments that cannot be accurately quantified, and rare mutations that are overwhelmed. In addition, almost all types of genetic variation, such as single nucleotide variants (SNV), small fragment insertion-deletion mutations (InDel), and large fragment structure variants (SV), can be detected and analyzed. Please feel free to share any comments, questions, or requests you might have. Our expert and experienced team will be in contact with you as soon as possible.
Case Studies
Case 1: The Genome of Antarctic krill
- Background
Antarctic krill (Euphausia superba) is a keystone species in the Southern Ocean ecosystem, serving as a fundamental link in the marine food web and contributing significantly to Earth's most abundant wild animal population.
- Challenges
The genome of Antarctic krill presents intriguing challenges due to its enormous size of 48.01 gigabases (Gb). This extensive genomic content, the largest reported among animal genomes, posed questions regarding the underlying genetic mechanisms responsible for such a large genome. Additionally, deciphering the genetic adaptations that enable Antarctic krill to thrive in the harsh and highly seasonal Antarctic conditions presented a significant scientific puzzle.
- Methods
Researchers undertook an ambitious genome sequencing project using a combination of cutting-edge techniques. Utilizing PacBio HiFi, NGS and Hi-C data, the team constructed a reference sequence of unparalleled accuracy for the Antarctic krill genome. This achievement yielded a genome assembly with a size of 48 Gb and a scaffold N50 of 1.08 Gb. A remarkable 66% of the genome sequence was anchored to 17 chromosomes, providing an intricate insight into the species' genetic structure.
- Results
The comprehensive genome assembly provided novel insights into the genetic adaptations of Antarctic krill. The study identified extensive repeat expansions, accounting for up to 92.45% of the genome, attributed to two explosive expansions of repetitive sequences. This unprecedented observation positioned Antarctic krill as a species with the highest repetitive sequence content among reported genomes. Additionally, the molecular architecture of the Antarctic krill circadian clock was revealed, shedding light on its ability to synchronize with the extreme variations in light and temperature. Expanded gene families associated with molting and energy metabolism were also discovered, further elucidating the species' resilience to the challenging Antarctic conditions.
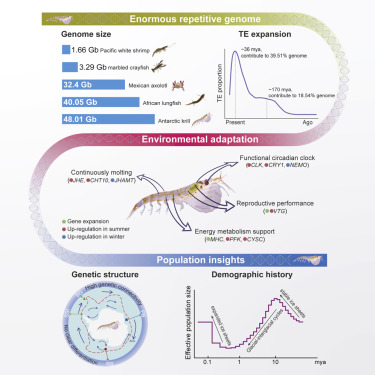 The Genome of Antarctic krill. (Shao et al., 2023)
The Genome of Antarctic krill. (Shao et al., 2023)
Case 2: Haplotype-resolved Genomes of Octoploid Strawberries
- Background
The strawberry (Fragaria spp.) has emerged as a fascinating model for unraveling the complexities of polyploid genome evolution and the rapid domestication of fruit crops. In this transformative case study, we delve into the genetic landscape of this remarkable plant, focusing on the wild octoploid species Fragaria chiloensis and Fragaria virginiana, which serve as the progenitors of our cultivated strawberries.
- Challenges
Exploring the genome of octoploid strawberries posed significant challenges due to their intricate genetic makeup and intricate evolutionary history. The task involved deciphering the distinct variation between species and within haplotypes, as well as disentangling the contributions of diploid ancestors.
- Methods
Our study employed a cutting-edge kmer-based approach to construct high-quality, chromosome-level haplotype genomes for F. chiloensis and F. virginiana. By leveraging a range of analytical methods, we meticulously retraced the footprints of the octoploid strawberries' diploid predecessors. Additionally, we conducted comprehensive sequencing of the diploid F. nipponica genome to further elucidate the genetic contributions of diploid species.
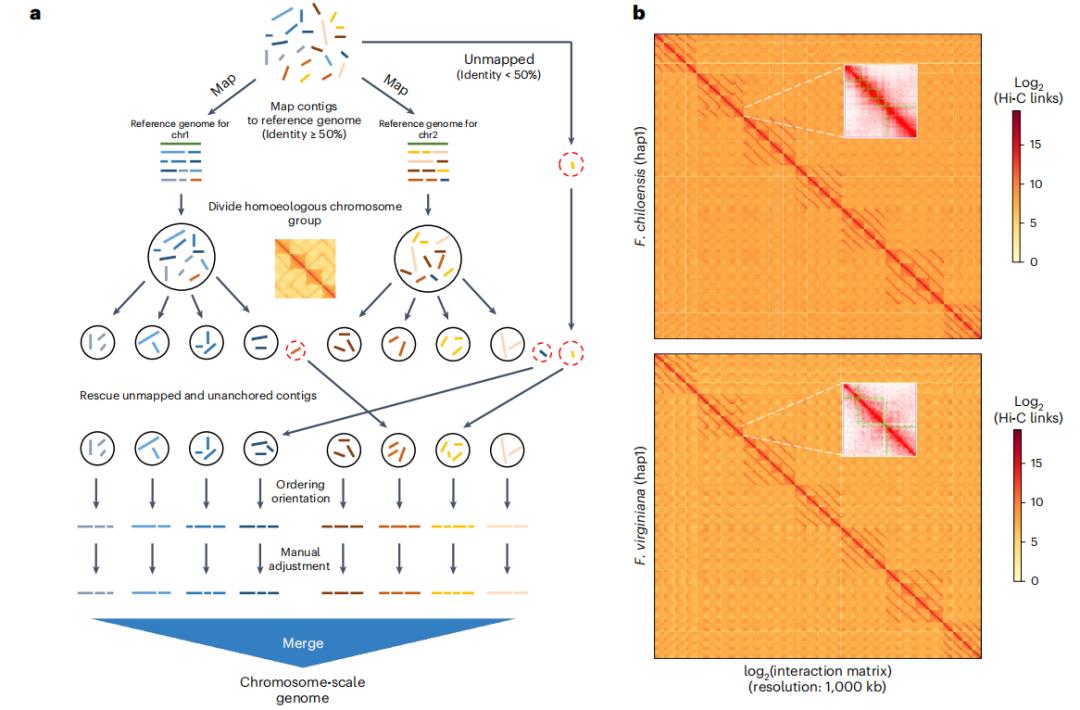 Haplotype-resolved Genomes of Octoploid Strawberries. (Jin et al., 2023)
Haplotype-resolved Genomes of Octoploid Strawberries. (Jin et al., 2023)
- Results
Our research has yielded groundbreaking insights into the evolutionary saga of octoploid strawberries. We redefined the four subgenomes that comprise their complex genome structure, shedding light on their intricate genetic architecture. Through a meticulous examination of homoeologous gene expression, we unveiled the phenomenon of subgenome dominance, homoeologous exchanges, and coordinated expression patterns. Notably, we identified crucial transcription factors that underwent significant expression shifts during the domestication process.
Case 3: T2T Reference Genome of Triploid Cavendish banana
- Background
Banana, a globally vital crop, plays a crucial role in sustaining human societies. Among the diverse banana cultivars, the Cavendish-type bananas, originating from the monospecific Musa acuminata (AAA), constitute nearly half of the world's banana production. Despite their significance, the absence of a high-quality, haplotype-resolved reference genome for banana cultivars has posed a major challenge.
- Challenges
The lack of a comprehensive reference genome has hindered deeper insights into the genetic makeup of banana cultivars, including variations, gene families, and key traits. Understanding these aspects is essential for molecular breeding, genetic enhancement, and further scientific exploration.
- Methods
In this groundbreaking study, we successfully decoded the telomere-to-telomere (T2T) and haplotype-resolved reference genome of the Cavendish banana variety. Employing state-of-the-art techniques, including HiFi sequencing, ONT sequencing, and Hi-C data, we achieved a comprehensive triploid reference genome with three distinct haploid assemblies.
- Results
Our research yielded three haploid assemblies of Cavendish banana, each approximately 477.16 Mb, 477.18 Mb, and 469.57 Mb in size. Despite sharing a monospecific origin, these assemblies exhibited remarkable differences and demonstrated low levels of sequence collinearity. Particularly notable were the identification of significant reciprocal translocations involving chromosomes 1, 4, and 7. An intriguing expansion of gene families associated with sucrose and starch metabolic processes, aromatic compound biosynthesis, and anther/pollen development came to light. This expansion of gene families could influence fruit quality, aroma, and reproductive traits. Moreover, the study revealed fewer resistance genes in Cavendish banana compared to M. acuminata, offering valuable insights for disease resistance research.
References
- Rehman, S. U., et al. (2021). "Whole-genome sequencing and characterization of buffalo genetic resources: recent advances and future challenges." Animals, 11(3), 904.
- Shao, Changwei, et al. "The enormous repetitive Antarctic krill genome reveals environmental adaptations and population insights." Cell 186.6 (2023): 1279-1294.
- Jin, Xin, et al. "Haplotype-resolved genomes of wild octoploid progenitors illuminate genomic diversifications from wild relatives to cultivated strawberry." Nature Plants (2023): 1-15.
- Huang, Hui-Run, et al. "Telomere-to-telomere haplotype-resolved reference genome reveals subgenome divergence and disease resistance in triploid Cavendish banana." Horticulture Research (2023): uhad153.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment