Inbreeding, also referred to as consanguinity or close-relative mating, involves the crossing of individuals with close genetic relationships to produce offspring. Depending on the degree of relatedness, inbreeding can be categorized into full siblings, half-siblings, and cousin inbreeding, with the most extreme form encompassing self-fertilization or backcrossing. Inbreeding results in increased homozygosity but may also induce inbreeding depression, thus it is frequently employed to stabilize desirable traits in production settings. It is important to note that the term inbred population in this context refers not to the inbreeding of original parental lines but rather to the inbreeding of progeny derived from different source parents (potentially distantly related) after hybridization, such as self-fertilization or backcrossing populations.
Such populations play a pivotal role in the construction of genetic linkage maps and the localization of Quantitative Trait Loci (QTL), and are thus also referred to as mapping populations. Based on the difficulty of obtaining these populations and their intended use, they can be classified into primary and secondary mapping populations.
Primary mapping populations are typically obtained through direct crosses, self-fertilization, or initial backcrossing procedures and are primarily utilized for preliminary QTL localization. These populations can be further classified into permanent populations (e.g., Doubled Haploids (DH), Recombinant Inbred Lines (RILs), Initial F2 populations) and temporary populations (e.g., F2, Backcross (BC) populations), depending on trait stability or genotype homozygosity.
Secondary mapping populations are developed from primary mapping populations through additional rounds of backcrossing and self-fertilization, resulting in populations with a more uniform genetic background, which facilitates finer QTL localization. Examples of secondary mapping populations include Single Segment Substitution Lines (SSSLs), Near Isogenic Lines (NILs), and Chromosome Segment Substitution Lines (CSSLs).
Furthermore, populations can be categorized based on the number of parental lines into biparental populations (e.g., F2, BC, RIL, NIL, DH) and multiparental populations (e.g., Nested Association Mapping (NAM), Multi-parent Advanced Generation Inter-Cross (MAGIC)). The following sections provide a brief overview of these commonly used population types.
The F2 population is formed by crossing two homozygous parents with different genotypes to produce an F1 generation, which is then selfed to create the F2 generation.
Derivative populations can be obtained from the F2 population through various methods. For instance, the F2:3 population consists of selfed progeny derived from individual F2 plants, maintained as distinct families. The IF2 population, also known as the permanent F2 population, is produced by random mating among recombinant inbred lines. This preserves the rich genetic variation of the F2 population while allowing for long-term conservation.
Backcross (BC) populations are generated by crossing F1 individuals with one of their parental lines. These populations are similar to F2 populations and are particularly suitable for creating segregating populations in the context of male sterility.
Recombinant Inbred Lines (RILs) are developed by continuous selfing or sibling mating of F2 individuals, leading to near-homozygous lines. The Single Seed Descent (SSD) method is a common approach for RIL development, though it requires multiple generations over several years.
| Advantages
| Disadvantages |
| - Homozygosity: Individual genotypes are homozygous, preventing segregation in subsequent generations. |
- Long Construction Period: The development of RILs, especially in achieving complete homozygosity, can be time-consuming. |
| - High Recombination: The recombination rate is higher than in F2 populations, leading to finer genetic map resolution. |
- Inability to Estimate Dominance Effects: Due to homozygosity, dominance effects cannot be assessed. |
Near Isogenic Lines (NIL)
Near Isogenic Lines (NILs) consist of individuals that are nearly identical in their chromosomal regions, except for a few loci. NILs are typically developed through multiple backcrosses, where a donor parent possessing a desired trait is crossed with a recurrent parent, followed by selfing after several backcross generations. This process results in lines with a genetic background similar to the recurrent parent but containing the trait of interest from the donor.
| Advantages |
Disadvantages |
| - Consistent Genetic Background: This uniformity allows for accurate and rapid molecular marker identification. |
- Long Construction Cycle: The development of NILs involves extensive backcrossing and selfing, requiring significant time and effort. |
| - Fine Mapping of QTLs: NILs enable the dissection of quantitative trait loci (QTL) into single Mendelian factors, facilitating precise mapping and positional cloning. |
- High Workload: The process is labor-intensive, demanding considerable resources. |
| - Epistatic Interactions: NILs provide an opportunity to study interactions between genes. |
|
NILs can be further categorized into Introgression Lines (ILs), Chromosome Segment Substitution Lines (CSSLs), and Single Segment Substitution Lines (SSSLs), based on the extent and specificity of the chromosomal regions substituted.
Doubled Haploids (DH)
Doubled Haploid (DH) populations are developed by inducing haploid individuals from F1 plants and subsequently doubling their chromosome number. Methods such as distant hybridization, androgenesis, gynogenesis, apomixis, or pollen culture can be used to generate haploids.
| Advantages |
Disadvantages |
| - Homozygous Plants: DH plants are homozygous, and their progeny remain true-breeding. |
- Limited Recombination: Recombination is restricted to the meiotic events during pollen formation, providing less recombination data. |
| - Repeated Trait Analysis: DH populations can be used for consistent experimentation on both qualitative and quantitative traits across multiple locations and years. |
- Difficulty in Culture: Some crops are challenging to culture, and DH lines may exhibit segregation distortion. |
| - Direct Representation of F1 Gametic Segregation: DH populations reflect the genetic segregation and recombination from the F1 generation, with mapping efficiency comparable to BC1. |
|
Limitations of Biparental Populations
All the aforementioned populations share several limitations:
Limited Allelic Variation: Biparental populations can only explore alleles at two loci, restricting the detection of genetic variance when both parents share the same allele.
Low Mapping Resolution: The limited number of recombination events during population development and the small number of alleles can reduce the resolution of QTL mapping.
To address these limitations, multiparental populations have been developed, involving more than two parental lines. Examples include Nested Association Mapping (NAM) populations and Multi-parent Advanced Generation Inter-Cross (MAGIC) populations. These populations combine the advantages of biparental and natural populations, offering both linkage analysis and association mapping capabilities.
Nested Association Mapping (NAM)
Nested Association Mapping (NAM) populations are created by crossing a common parental line (CP) with multiple diverse parental lines, followed by extensive selfing to produce a series of recombinant inbred line (RIL) populations. Each RIL population contains the CP, resulting in a large, diverse genetic population.
| Advantages |
Disadvantages |
| - Fixed Common Parent: The use of a single common parent simplifies the analysis of the population structure. |
- Time-Consuming Construction: The development of NAM populations requires significant time and effort. |
| - Ease of Analysis: The population structure is easier to analyze compared to more complex multiparental populations. |
|
| - Combines Strengths of Biparental and Natural Populations: NAM populations integrate the benefits of both biparental and natural populations, offering the advantages of linkage analysis and association mapping. |
|
The first NAM population was developed using the maize inbred line B73 as the common parent, crossed with 25 different inbred lines, resulting in 25 RIL populations comprising approximately 5000 lines. This population has been successfully utilized for studies on traits such as flowering time and resistance to leaf blight in maize.
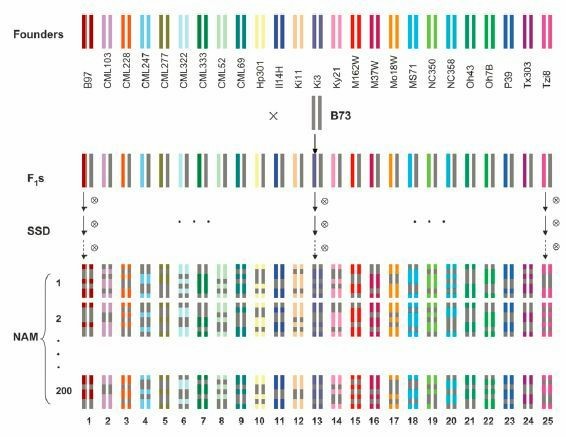 Figure 1. Diagram of genome reshuffling between 25 diverse founders and the common parent and the resulting 5000 immortal genotypes. (Yu, J. et al., 2008)
Figure 1. Diagram of genome reshuffling between 25 diverse founders and the common parent and the resulting 5000 immortal genotypes. (Yu, J. et al., 2008)
Multiparent Advanced Generation Inter-Cross (MAGIC)
Multiparent Advanced Generation Inter-Cross (MAGIC) populations are created through a complex series of crosses involving multiple parental lines. Initially, pairs of parental lines are crossed to produce F1 hybrids. These F1 hybrids are then crossed again in pairs to create double-cross progeny. The double-cross progeny are further crossed, resulting in offspring derived from eight different parents. This crossing process can continue to incorporate additional parental lines. Finally, the complex hybrid progeny are selfed or doubled haploids are produced, generating a series of recombinant inbred line (RIL) populations or doubled haploid (DH) populations.
| Advantages |
Disadvantages |
| - Simultaneous Mapping of Multiple Major QTLs: MAGIC populations allow the identification of multiple quantitative trait loci (QTLs) simultaneously. |
- Time-Consuming and Labor-Intensive Construction: The development process is complex and requires significant time and resources. |
| - No Decline in Polymorphism or Hybrid Vigor: Due to the use of multiple parental lines, MAGIC populations maintain genetic diversity and avoid inbreeding depression. |
- Complex Population Structure: The intricate crossing scheme can make population structure difficult to manage and analyze. |
| - Enhanced Hybrid Vigor: The introduction of diverse genetic backgrounds can lead to pronounced hybrid vigor. |
|
The first MAGIC population was developed in Arabidopsis thaliana, derived from approximately 19 different Arabidopsis accessions. This population, consisting of 527 recombinant inbred lines, has been utilized for fine mapping of QTL traits in Arabidopsis.
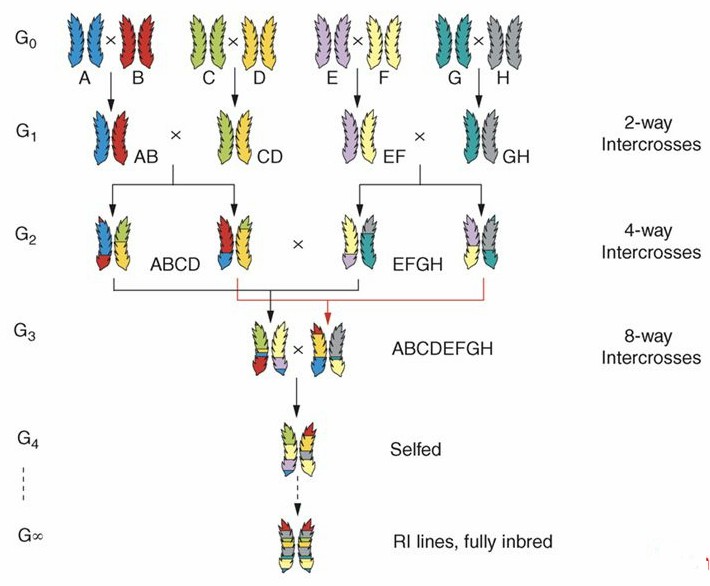 Figure 2. MAGIC populations are created by inter-crossing n lines for n/2 generations until all founders are combined with equal proportions in the inter-crosses, RILS may be derived immediately or additional rounds of intermating. (Cavanagh et al., 2008)
Figure 2. MAGIC populations are created by inter-crossing n lines for n/2 generations until all founders are combined with equal proportions in the inter-crosses, RILS may be derived immediately or additional rounds of intermating. (Cavanagh et al., 2008)
Natural Populations
Natural populations, unlike biparental or multiparental populations, do not suffer from the constraints of limited parental genotypes, thereby offering a broader spectrum of genetic information. These populations are typically maintained across generations through natural mechanisms, which, however, may limit the precision of mapping efforts. While designed mating populations, such as those derived from biparental and multiparental crosses, are primarily employed for linkage analysis, natural populations are predominantly utilized for association mapping.
Natural populations are composed of a diverse array of genotypes, originating from various sources, including:
Germplasm Resources: These represent a broad spectrum of genetic diversity available for study and utilization.
Modern Commercial Varieties and Their Progenitors: These include contemporary cultivars developed through advanced breeding techniques.
Mutant Libraries: Collections derived from a single parent subjected to mutagenesis, facilitating the study of specific genetic mutations.
Landraces/Farmers' Varieties: Traditional varieties developed and maintained by farmers over generations, often exhibiting unique genetic traits.
Wild Relatives: Species closely related to cultivated crops, often harboring desirable traits for breeding.
The diversity within natural populations, particularly concerning target traits, provides a rich resource for association mapping. However, to avoid confounding effects, it is critical to ensure that the variability in non-target traits remains as random as possible.
Genetic Mating Populations
Genetic mating populations are meticulously designed constructs in genetic research, engineered through specific mating strategies to serve targeted experimental purposes. These populations include, but are not limited to, designs such as Diallel Crosses, North Carolina Design (NCD) I-III, Triple Test Crosses (TTC), and Split Triple Test Crosses (sTTC). These structured populations are crucial for dissecting genetic variance, particularly focusing on the additive components.
The primary objective of genetic mating designs is to precisely estimate the additive genetic variance within a population. This is achieved through the use of well-defined reference populations, which are drawn from specific genetic groups to ensure accurate variance estimation. These populations are referred to as reference or base populations.
Diallel Crosses
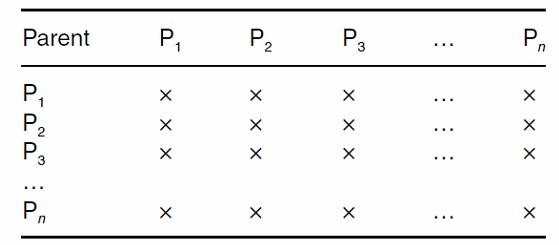
A specific and widely used design is the Diallel Cross, which involves crossing multiple parental lines in all possible combinations. The resultant F1 hybrids from these crosses form a diallel population, designed to maximize the expression of genetic diversity. This method allows for comprehensive analysis of the genetic architecture underlying complex traits by assessing the performance of each hybrid combination.
Genetic Mating Designs in Breeding Research
Diallel Cross Designs
Diallel cross designs are foundational in the analysis of genetic variance, providing a comprehensive framework to evaluate genetic interactions among parental lines. These designs can be categorized into four primary types:
Full Diallel: This design involves all possible pairwise crosses between a set of parental lines, including self-pollinations, providing a complete representation of genetic interactions.
Full Diallel without Parents: This variant excludes self-pollinations, thereby eliminating the diagonal elements in the resulting matrix, which represent self-crosses.
Half Diallel: In this approach, only one set of reciprocal crosses is performed, typically either the upper or lower triangular matrix, excluding self-pollinations. This design reduces redundancy and focuses on one direction of crossing.
Half Diallel without Parents: Similar to the half diallel, this design excludes self-pollinations and retains either the upper or lower triangular matrix of crosses. It is frequently employed in breeding programs to simplify the analysis and reduce experimental load.
North Carolina Design (NCD)
The North Carolina Design (NCD) is a set of experimental designs used to estimate the components of genetic variance. It is particularly valuable in studying hybrid vigor (heterosis) and in combining ability studies. The NCD populations are constructed through a variety of crossing schemes, including random mating among F2 individuals, backcrossing to parental lines, or crossing among different inbred lines.
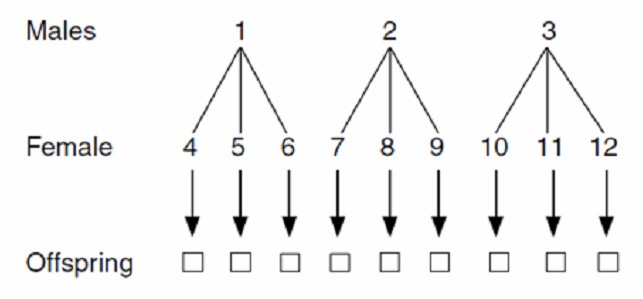
NCD I: Two-Factor Nested Mating Design
In the NCD I design, a random sample of individuals from the reference population is selected to serve as male parents. For each male parent, a random set of individuals is chosen as female parents, resulting in a nested mating structure. This design allows for the estimation of both additive and non-additive genetic variance components, and is widely used in the study of heterosis and genetic diversity.
NC II: Two-Factor Crossbreeding Design
The NC II design, or Two-Factor Crossbreeding Design, involves the systematic crossbreeding of two distinct sets of parental lines. In this design, each male parent is crossed with every female parent from a separate group, resulting in a comprehensive set of crosses. This design facilitates the evaluation of both additive and non-additive genetic effects by considering all possible combinations between the two sets of parents.
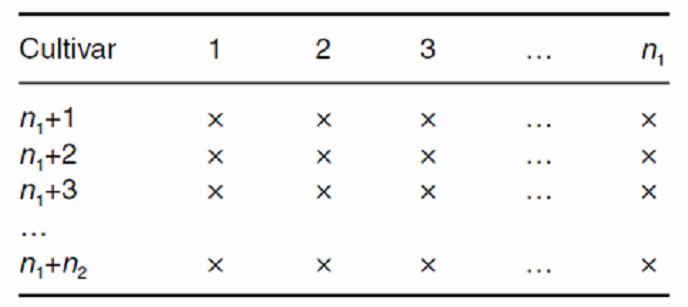
NC III: Backcross Mating Design
The NC III design, or Backcross Mating Design, is employed to assess genetic variation through a series of backcrosses. In this design, individuals from the F2 generation, derived from a cross between two pure lines (P1 and P2), are randomly selected and subsequently backcrossed with the original parental lines (P1 and P2). This process generates 2n hybrid combinations, with each combination being evaluated based on r progeny. This design allows for the estimation of additive and dominance effects by comparing progeny performance across various genetic backgrounds.
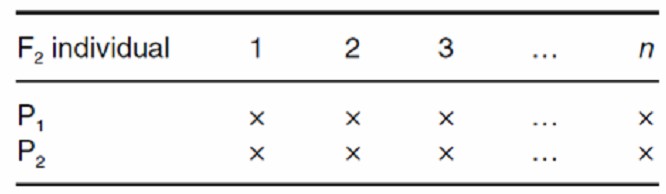
TTC
Test Cross (TC)
Test Cross populations (TC) involve crossing F1 progeny with recessive or double-recessive parental lines. This design is used to test hybrid vigor or to assess genetic compatibility. By analyzing the performance of F1 individuals when crossed with known recessive lines, researchers can infer the genetic contributions of different parental lines and evaluate the interaction of genetic factors.
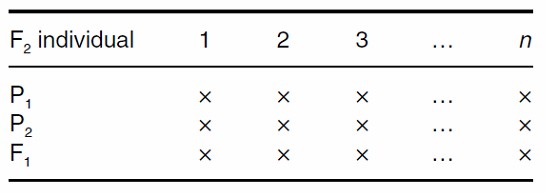
Triple Test Cross (TTC)
The Triple Test Cross (TTC) design extends the test cross approach by including multiple backcrosses. In this design, more than 20 single plants from the F2 generation, derived from crosses between parental lines, are selected and crossed with both original parents and F1 progeny. This results in a more detailed assessment of genetic interactions and enables the evaluation of genetic effects with higher precision.
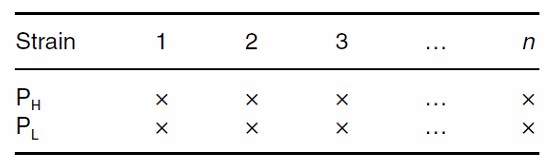
Simplified Triple Test Cross (sTTC)
The Simplified Triple Test Cross (sTTC) design streamlines the traditional TTC approach. This design maintains the rigorous testing of dominance effects but is more cost-effective and efficient. Unlike the full diallel crosses, the sTTC approach typically focuses on a single phenotype per study, allowing for a more precise estimation of breeding levels. This simplification makes the sTTC a valuable tool for genetic analysis, particularly in contexts where resource constraints are a consideration.
The relationships between various genetic mating designs are summarized as follows:
NC II and NC III designs focus on crossbreeding and backcrossing strategies to evaluate genetic variance and compatibility.
TC and TTC designs are used for testing hybrid vigor and genetic interactions through specific mating schemes.
sTTC represents a more streamlined approach to the TTC, balancing cost and analytical precision.
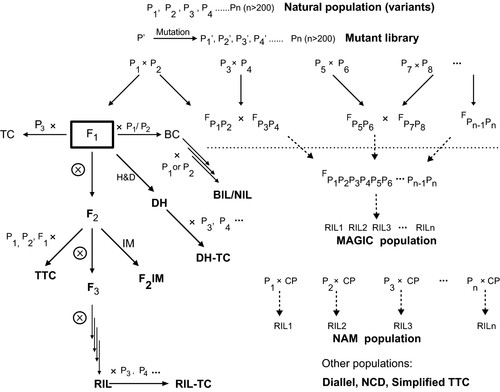 Figure 3. Plant populations and their relationships. (Zou et al., 2016)
Figure 3. Plant populations and their relationships. (Zou et al., 2016)
CD Genomics has successfully developed an integrated hardware and software system for breeding research. This system encompasses a full suite of tools, including phenotype data systems, genotype big data systems, germplasm management systems, genomic/multi-omics databases, and a genome-wide selection (GWS) prediction platform. This comprehensive system is designed to provide research clients with end-to-end breeding solutions, streamlining the process from data collection and information management to intelligent analysis, thereby ensuring systematic, scientific, and efficient breeding practices.
References
- Yu, J., Holland, J. B., McMullen, M. D., & Buckler, E. S. (2008). Genetic design and statistical power of nested association mapping in maize. Genetics, 178(1), 539-551. https://doi.org/10.1534/genetics.107.074245
- Zou, C., Wang, P., & Xu, Y. (2016). Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnology Journal, 14(10), 1941-1955. https://doi.org/10.1111/pbi.12559
- Cavanagh, C., Morell, M., Mackay, I., & Powell, W. (2008). From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Current Opinion in Plant Biology, 11(2), 215-221. https://doi.org/10.1016/j.pbi.2008.01.002
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.