
 Sample Submission Guidelines
Sample Submission GuidelinesCD Genomics is providing PacBio SMRT sequencing to complement our NGS facility. By taking advantage of the long-read and single molecular sequencing capability developed by PacBio, we are proud to offer advanced genome de novo assembly solutions and full-length gene/transcript sequencing strategy to suit your project needs.
Introduction of PacBio SMRT Sequencing
Single Molecular Real-Time (SMRT) sequencing employs a specialized flow cell with many thousands of individual picolitre wells with transparent bottoms — zero-mode waveguides (ZMW). The polymerase is fixed to the bottom of the well and allows the DNA strand to progress through the ZMW. As a result, the system can focus on a single molecular. SMRT sequencing allows for real-time imaging of fluorescently tagged nucleotides that are synthesized along with individual DNA template molecules. The sequencing reaction ends when the template and polymerase dissociate. The average read length from the PacBio instrument is approximately 2 kb, and some reads may be over 20 kb. Longer reads are especially useful for de novo assemblies of novel genomes that can span many more repeats and bases.
Highly repetitive elements found in both eukaryotic and prokaryotic genomes pose a challenge for genome assembly and make the detailed study of repetitive sequences difficult. Long-read sequencing delivers reads in excess of several or dozens of kilobases (kbs), which can span complex or repetitive regions with a single continuous read, allowing for the resolution of these large structural features. Besides considerably longer and highly accurate DNA sequences from individual unamplified molecules, it can also exhibit where methylated bases occur, thereby providing functional information about DNA methyltransferases encoded by the genome. PacBio SMRT sequencing has unique advantages in studies of de novo genomics, metagenomics, transcriptomics and epigenetics.
Advantages of Our PacBio SMRT Sequencing Service
- Longest average read lengths
- Highest consensus accuracy
- Uniform coverage
- Simultaneous epigenetic characterization
- Single-molecule resolution
- Rapid and affordable
Application of PacBio SMRT Sequencing
- High-quality de novo genome assembly: PacBio's long-read advantage makes it the primary technology for high-quality de novo genome assembly. Its contiguous long reads help overcome challenges such as repetitive regions, base composition bias, and structural rearrangements in the genome.
- Exploration of genomic regions: PacBio sequencing can aid in exploring genomic regions previously difficult to sequence due to long repetitive sequences, strong base composition bias, or structural rearrangements.
- Iso-Seq transcript sequencing: PacBio's Iso-Seq technology is used for analyzing full-length mRNA transcripts without assembly. This helps gain insights into the panorama of gene expression, particularly for complex transcript rearrangements.
- Long amplicon sequencing: PacBio sequencing can be employed for sequencing long amplicons, suitable for detecting long DNA fragments such as full-length gene amplicons and genomic rearrangements.
- Bacterial DNA base modification studies: PacBio sequencing technology can be utilized for studying bacterial DNA base modifications, such as N6-methyladenine (m6A) and N4-methylcytosine (m4C), to understand their impacts on processes like gene expression, gene silencing, and DNA replication.
- Epigenomic Studies: PacBio SMRT (Single Molecule, Real-Time) sequencing technology presents a unique application value in the field of epigenomics through its ability to accurately sequence DNA methyl modifications directly without bisulfite conversion. This bears a significant potential to expound on directions for studies in this discipline, thereby exploring the mysteries of complex cellular processes.
- Diversity and Functional Research in Microbial Communities: PacBio SMRT sequencing offers resolutions to the challenges in full-genome sequencing of pathogens and in the taxonomic identification of hard-to-culture microorganisms. Its capacity for long-read sequencing enables comprehensive genomic surveillance of microbial communities, thereby providing crucial insights into their diversity and functional roles in various ecosystems.
Our PacBio SMRT Sequencing services
We utilize the advanced PacBio SMRT instruments (PacBio SR II and PacBio Sequel) for several research purposes including whole-genome de novo genome assembly, full-length target sequencing, metagenomics studies, full-length transcripts sequencing, and genome-wide DNA modification analysis. Our highly experienced expert team executes quality management following every procedure to ensure confident and unbiased results.
-
Long-Read metagenomic sequencing
- Metagenomics, defines as the direct genetic analysis of genomes contained within an environmental sample. This field was initially launched off by the cloning of environmental DNA, followed by functional expression screenings and soon bolstered by the sequencing of environmental DNA. Long-read metagenomic sequencing, realized by platforms such as PacBio SMRT, allows for direct genetic analysis of diverse microbial communities extracted from various environmental samples. This technology facilitates the generation of longer read lengths, thereby triumphing over the repetitive elements seen in bacterial genomes. Consequently, it offers comprehensive insights into microbial functionalities, pathways, and the integral relationships they maintain with their dwelling habitats.
The applications of long-read metagenomic sequencing are broad and robust, spanning across diverse fields such as environmental science, medical biology, agriculture, as well as biotechnology. These applications provide profound avenues for understanding the diversity inherent in microbial communities, ecosystem dynamics, the mechanisms of diseases, and the process of bioprospecting. In conclusion, the PacBio SMRT technology serves as an influential and potent tool to expedite research progress and practical applications across a myriad of academic and industrial fields. The potency of this technology will continually be expanded and refined in the years to come.
- Metagenomics, defines as the direct genetic analysis of genomes contained within an environmental sample. This field was initially launched off by the cloning of environmental DNA, followed by functional expression screenings and soon bolstered by the sequencing of environmental DNA. Long-read metagenomic sequencing, realized by platforms such as PacBio SMRT, allows for direct genetic analysis of diverse microbial communities extracted from various environmental samples. This technology facilitates the generation of longer read lengths, thereby triumphing over the repetitive elements seen in bacterial genomes. Consequently, it offers comprehensive insights into microbial functionalities, pathways, and the integral relationships they maintain with their dwelling habitats.
-
Bacterial whole genome de novo sequencing
- Bacterial whole genome de novo sequencing entails the comprehensive reconstruction of a bacterium's genetic blueprint, devoid of reliance on a reference genome. This methodology affords a nuanced comprehension of bacterial genetic variability, evolutionary dynamics, and functional components, encompassing pivotal genes implicated in virulence, antimicrobial resistance, and adaptive responses to environmental cues. The spectrum of applications for bacterial whole genome de novo sequencing spans diverse domains, ranging from clinical microbiology, where it facilitates the prompt diagnosis and surveillance of infectious diseases and epidemics, to environmental microbiology, enabling the in-depth exploration of microbial consortia and their ecological interplay.
Single Molecule, Real-Time (SMRT) sequencing can generate long read lengths (average >15,000 bp, with some sequences exceeding 100,000 bp) and boasts the highest consistency and accuracy. This makes SMRT sequencing remarkably useful for de novo genome assembly. One of the key advantages of SMRT's extensive read lengths is the ability to overcome obstacles often encountered in genomics research, such as GC bias and high replication rates in bacterial genomes. Consequently, SMRT sequencing often results in the assembly of bacterial genomes into single contigs, circumventing regions of ambiguity and resultant multiple possibilities that can complicate the assembly process.
- Bacterial whole genome de novo sequencing entails the comprehensive reconstruction of a bacterium's genetic blueprint, devoid of reliance on a reference genome. This methodology affords a nuanced comprehension of bacterial genetic variability, evolutionary dynamics, and functional components, encompassing pivotal genes implicated in virulence, antimicrobial resistance, and adaptive responses to environmental cues. The spectrum of applications for bacterial whole genome de novo sequencing spans diverse domains, ranging from clinical microbiology, where it facilitates the prompt diagnosis and surveillance of infectious diseases and epidemics, to environmental microbiology, enabling the in-depth exploration of microbial consortia and their ecological interplay.
-
Fungal whole genome de novo sequencing
- Fungi occupy a pivotal niche within the biosphere, exerting profound impacts on both ecological balance and human affairs, owing to their significant medical and economic ramifications. Unraveling the complete genome of fungi lays a robust groundwork for mycological investigations, fostering deeper insights into fungal biodiversity, growth dynamics, nutritional requirements, physiological attributes, genetic underpinnings, metabolic pathways, and ecological roles. Fungal whole genome de novo sequencing entails the meticulous deciphering of the entire genetic repertoire of a fungus, independent of any reference genome. This approach furnishes holistic perspectives on fungal genomics, evolutionary trajectories, and functional attributes, encompassing genes implicated in pathogenesis, environmental adaptability, and biosynthesis of secondary metabolites.
Through the use of long reads achieved via Single Molecule, Real-Time (SMRT) sequencing on the PacBio system, we can generate comprehensive de novo assemblies for fungal genomes. These assemblies approach or attain zero gaps or N-based errors with a contig N50 >1 Mb and a high degree of accuracy at 99.999%. This innovative technique presents a significant stride towards the improvement of fungal genome studies.
- Fungi occupy a pivotal niche within the biosphere, exerting profound impacts on both ecological balance and human affairs, owing to their significant medical and economic ramifications. Unraveling the complete genome of fungi lays a robust groundwork for mycological investigations, fostering deeper insights into fungal biodiversity, growth dynamics, nutritional requirements, physiological attributes, genetic underpinnings, metabolic pathways, and ecological roles. Fungal whole genome de novo sequencing entails the meticulous deciphering of the entire genetic repertoire of a fungus, independent of any reference genome. This approach furnishes holistic perspectives on fungal genomics, evolutionary trajectories, and functional attributes, encompassing genes implicated in pathogenesis, environmental adaptability, and biosynthesis of secondary metabolites.
-
Full-length transcripts sequencing (Iso-Seq)
- Full-Length Transcript Sequencing (Iso-Seq) stands at the forefront of molecular biology techniques, reshaping our comprehension of gene expression. In contrast to conventional RNA sequencing methods, prone to generating fragmented sequences, Iso-Seq captures complete transcripts from initiation to termination. This capability allows for precise identification and quantification of alternatively spliced isoforms and rare transcripts, presenting a holistic depiction of the transcriptome landscape.
Through the revelation of transcript full-length structures, Iso-Seq facilitates the uncovering of new genes, isoforms, and non-coding RNAs, unveiling previously uncharted territories of gene regulation and function. Its versatile applications span diverse domains, encompassing developmental biology, cancer research, and neuroscience, furnishing indispensable insights into the intricate mechanisms orchestrating cellular processes.
In comparison with the Illumina platform, PacBio analyses are capable of easily detecting extremely long multi-exonic RNA molecules, denoted as complex transcripts, and expose various novel transcriptional overlaps between parallel adjacent and distant genes. This development assists in enhancing the potential for investigating the integrated control of gene expression and replication across the entire genomic network.
- Full-Length Transcript Sequencing (Iso-Seq) stands at the forefront of molecular biology techniques, reshaping our comprehension of gene expression. In contrast to conventional RNA sequencing methods, prone to generating fragmented sequences, Iso-Seq captures complete transcripts from initiation to termination. This capability allows for precise identification and quantification of alternatively spliced isoforms and rare transcripts, presenting a holistic depiction of the transcriptome landscape.
-
Human Whole Genome PacBio SMRT Sequencing
- On February 15 and 16, 2001, the journals Nature and Science respectively published the comprehensive sequence data collated by the Human Genome Project and Celera Genomics Project. Human genome sequencing denotes the elaborate process of mapping all DNA sequences within the human genome. The aim of this undertaking is to decode the components, structure, and functionality embedded in the human genome, in addition to unravelling correlations between human genes, health, and disease.
Human Whole Genome PacBio SMRT Sequencing stands as a cutting-edge genomics method, providing profound insights into the intricate nature of the human genome. Driven by PacBio's Single Molecule, Real-Time (SMRT) sequencing technology, this advanced approach yields lengthy, precise reads, ensuring thorough exploration of genomic segments with utmost accuracy. With its ability to capture structural variations, repetitive sequences, and complex genomic regions with remarkable detail, PacBio SMRT sequencing empowers researchers to unravel genetic diversity, evolutionary patterns, and disease mechanisms to an unprecedented extent. Its broad spectrum of applications spans diverse research domains, encompassing population genetics, personalized medicine, and disease diagnosis, thus fostering groundbreaking discoveries in human biology and healthcare.
- On February 15 and 16, 2001, the journals Nature and Science respectively published the comprehensive sequence data collated by the Human Genome Project and Celera Genomics Project. Human genome sequencing denotes the elaborate process of mapping all DNA sequences within the human genome. The aim of this undertaking is to decode the components, structure, and functionality embedded in the human genome, in addition to unravelling correlations between human genes, health, and disease.
-
Full-Length 16S/18S/ITS Amplicon Sequencing
- Given the progression of sequencer technology, 16S/18S/ITS rRNA gene sequencing has become the optimal tool for studying bacterial and fungal taxonomy and molecular evolution. Unlike mainstream, short read sequencing methods, which produce fragmental sequences of the objective genes, full-length amplicon sequencing captures the total length of the 16S, 18S, or ITS regions. Offering a more accurate depiction of microbial diversity, it enables accurate taxonomic categorization and species identification. By securing complete amplicon sequences, investigators can delve deeper into microbial ecology, evolution, and interactions within intricate ecosystems. Moreover, full-length amplicon sequencing promotes the discovery of novel taxa and functional genes, thereby advancing our comprehension of microbial communities and their role in various environments.
Sample requirements
| Service | Sample Type | Recommended Quantity | Minimum Quantity | Minimum Concentration |
|---|---|---|---|---|
| Whole Genome Sequencing | Genomic DNA | ≥ 3 µg | 80 ng/µL | |
| Full-Length 16S/18S/ITS Amplicon Sequencing |
Genomic DNA Tissue Thallus Interstitial Fluid Environmental Samples Water filter membrane |
≥ 500ng 1-3g 5 g 3-5 mL 3-5g 3 |
1 g 3 g 1 mL 1 g 1 |
10 ng/µL |
| Long-Read Metagenomic Sequencing | Genomic DNA Tissue Interstitial Fluid Environmental Samples Water filter membrane |
≥ 2 μg 2 g 6-10 mL, sediment 2g 6g 6 |
1 g 2 mL 2 g 2 |
30ng/µL |
| Iso-Seq | Total RNA | ≥ 2 μg | 600 ng | 30 ng/µL |
Analysis Pipeline
Our bioinformatics pipeline includes de novo assembly, base modification detection, single molecule consensus generation, transcript analysis, amplicon analysis, sequence alignment with variant detection. And more data mining are available based on your specific needs.
Supported by our experienced scientists and advanced platforms, CD genomics can assist you in the studies on genomics, transcriptomics, epigenomics, microbial genomics, and single-cell sequencing. with unmatched read lengths, uniform coverage, and high accuracy. If you have additional requirements or questions, please feel free to contact us.
References:
- PacBio's website
- Rhoads A, Au K F. PacBio sequencing and its applications. Genomics, Proteomics and Bioinformatics, 2015, 13(5): 278-289.
1. What are the differences between PacBio RS II and PacBio Sequel?
RS II system was released by PacBio in 2013, based on the SMRT technology, of which the average read length reaches 10 kb with the longest one beyond 20 kb. Sequel is the newest platform of PacBio that is also based on SMRT technology but has a large increase in throughput and data quality.
Table 1. The comparison between PacBio RS II and Sequel.
| Parameters | RS II | Sequel |
|---|---|---|
| principles | SMRT | SMRT |
| Average read length | 10-15kb | 8-12kb |
| Data sizes/SMRTcells | 500Mb-1Gb (750Mb-1.5Gb) | 5-10Gb |
| Max Output/Run | 18Gb (24Gb) | 160Gb |
| Run time/SMRTcell | 0.5-6 hours | 0.5-6 hours |
| Multiplex Amplicons | 384 | 1536 |
2. What is size selection?
PacBio allows for the sequencing of full-length cDNA libraries at single-molecule resolution. However, most transcripts are 1-1.5kb. Longer transcripts are hard to detect due to low abundance, amplification bias, and preferential loading of smaller SMRTbell constructs. Therefore, size selection is a powerful way to dramatically increase the number of transcripts>1.5kb. This is especially essential for transcripts>3kb. As for degraded samples, shearing is not necessary, which may further reduce library insert size, and size selection is important to remove shorter fragments that will be much less beneficial in assembly. Size selection can be performed with agarose gels or BluePippin system. Additionally, size selection can be used for whole-genome de novo sequencing .
3. The requirements of DNA samples.
In general, the following precautions need to be taken when submitting DNA samples:
- Make sure the purification and integrity of the DNA samples.
- Avoid overdrying of DNA.
- Avoid ethidium/UV based visualization when using gel purification.
- Avoid overheating and vortexing DNA.
- Undergo a minimum of freeze-thaw cycles.
- Double-stranded DNA.
Table 2. The estimated yield and recommended amounts of DNA samples.
| Library insert size | Recommended quantity for submission | Min concentration required (Post-shearing) | Est. Total yield (range) | |
|---|---|---|---|---|
| Min | Max | |||
| 250bp | 600ng | 250ng | 60Gb | 125Gb |
| 500bp | 600ng | 250ng | 10 Gb | 20Gb |
| 1kb | 1.2μg | 500ng | 90Gb | 180Gb |
| 2kb | 1.2μg | 500ng | 45Gb | 90Gb |
| 5kb | 2.4μg | 1μg | 45Gb | 91Gb |
| 10kb | 2.4μg | 1μg | 20Gb | 45Gb |
| 10kb (AMPure kit) | 10μg | 5μg | 90Gb | 182Gb |
| 20kb (AMPure kit) | 15μg | 5μg | 45Gb | 91Gb |
| 20kb (BluePippin kit) | 15μg | 5μg | 9Gb | 18Gb |
4. The requirements for RNA samples.
Please provide integrated and purified RNA samples (RNA ≥ 5 µg; concentration ≥ 300 ng/µl) for full-length transcripts sequencing.
Using PacBio sequencing to investigate the effects of treatment with lactic acid bacteria or antibiotics on cow endometritis
Journal: Microbiome
Impact factor: 16.837
Published: 22 January 2021
Backgrounds
This article investigates the intra-lineage diversities of pelagic bacterioplankton assemblages in 11 deep freshwater lakes in Japan and Europe. Despite the cosmopolitan nature of bacterioplankton lineages in freshwater ecosystems, their microdiversity and phylogeography remain unclear. The study utilizes long-read amplicon sequencing targeting nearly full-length 16S rRNA genes and adjacent ribosomal internal transcribed spacer sequences to address these gaps in understanding.
Methods
- Nine Japanese and two European perialpine lakes
- DNA extraction
- Polymerase chain reaction (PCR)
- Library preparation
- Full-Length 16S/ITS Amplicon Sequencing
- PacBio RSII sequencing
- Analysis of sequencing outputs
- Hierarchical clustering
- Metagenomic read mapping
Results
1. Methodological performance and limitations
The study evaluates the performance and limitations of long-read sequencing platforms, focusing on read accuracy, throughput, and community composition discrepancies. Targeting 16S rRNA gene and ITS sequences while excluding the adjacent 23S rRNA gene helps maintain read accuracy. Validation using SNPs identified from ASVs and metagenomic read mapping demonstrates high reproducibility. However, low read throughput hampers analysis performance, limiting reads assigned to ASVs and hindering alpha diversity estimates. Primer specificity and rRNA gene operon connectivity contribute to discrepancies in community composition compared to short-read sequencing. Despite limitations, long-read amplicon sequencing shows promise for high-resolution phylogenetic profiling of environmental microbial assemblages.
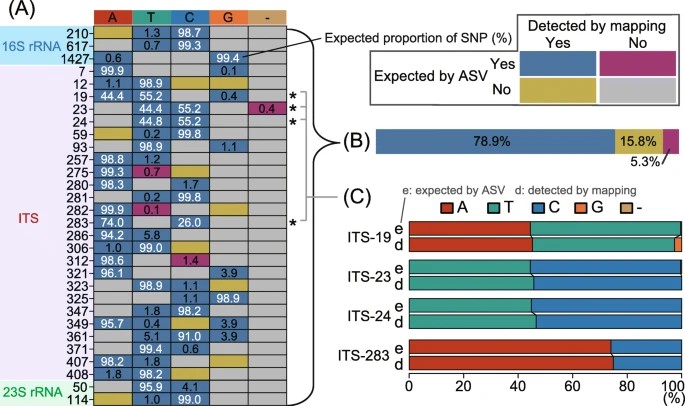 Figure 1. Comparison of single-nucleotide polymorphisms (SNPs) expected based on amplicon sequence variants (ASVs) and those detected through metagenomic read mapping of CL500-11 sequences from Lake Biwa.
Figure 1. Comparison of single-nucleotide polymorphisms (SNPs) expected based on amplicon sequence variants (ASVs) and those detected through metagenomic read mapping of CL500-11 sequences from Lake Biwa.
 Figure 2. Schematic illustration of mismatches and gaps found in the 16S rRNA gene and internal transcribed spacer (ITS) sequences of the CL500-11 lineage.
Figure 2. Schematic illustration of mismatches and gaps found in the 16S rRNA gene and internal transcribed spacer (ITS) sequences of the CL500-11 lineage.
2. Phylogenetic composition of long-read amplicons
The analysis identified 742 ASVs, revealing diverse populations within lineages with high sequence identity. Dominant phyla varied between epilimnion and hypolimnion samples. Further analysis focused on 11 OTUs termed "dominant lineages," representing 85.4% of total read abundance, across Chloroflexi, Alphaproteobacteria, and Actinobacteria.
3. Intra-lineage microdiversity and phylogeographic patterns among lakes
The study investigates the intra-lineage microdiversity and phylogeographic patterns among bacterial plankton communities in freshwater lakes. By analyzing nearly complete 16S rRNA gene sequences, it reveals diverse communities within high sequence similarity. Dominant lineages exhibit varying degrees of microdiversity, with a few ASVs dominating each sample. Cluster analysis based on ASV composition shows the influence of region and environment on population structure. Migration between lakes is limited by distance, but environmental factors and genetic drift also play significant roles. Temporal analysis indicates stability over time, with dominant ASVs shared across different years in Lake Biwa, suggesting temporal consistency. However, further investigation across seasons and years is warranted for comprehensive understanding.
 Figure 3. Clustering of samples based on the Bray–Curtis dissimilarity of amplicon sequence variant composition generated by averaging the values for the 11 most dominant lineages.
Figure 3. Clustering of samples based on the Bray–Curtis dissimilarity of amplicon sequence variant composition generated by averaging the values for the 11 most dominant lineages.
4. Microdiversity and phylogeographic patterns of the most abundant hypolimnetic lineage (CL500-11)
The study examines the microdiversity and phylogeographic patterns of CL500-11, a predominant bacterioplankton lineage inhabiting deep freshwater lake hypolimnia. It identifies distinct ASVs dominating Japanese and European lakes, showing genetic differentiation in the 16S rRNA gene and ITS region between these populations. Surprisingly, microdiversification within Japanese lakes is limited, challenging assumptions about factors influencing hypolimnetic lineage diversification. Comparison with previous metagenome analyses suggests regional genetic clustering within CL500-11, but intercontinental connectivity is observed between Europe and North America. The study highlights the need for further exploration of CL500-11's genetic diversity across different regions and timeframes.
 Figure 4. Distribution of pairwise Bray–Curtis dissimilarity for amplicon sequence variant (ASV) compositions among the nine Japanese lakes.
Figure 4. Distribution of pairwise Bray–Curtis dissimilarity for amplicon sequence variant (ASV) compositions among the nine Japanese lakes.
Conclusion
In conclusion, this novel approach supplements the limited resolution of short-read amplicon sequencing and the restricted sensitivity of metagenome assembly-oriented strategies, providing more insightful perspectives into the intricate ecological processes that govern the ubiquity of freshwater bacterioplankton lineages. Nevertheless, it should be noted that the relatively low read throughput of the method serves as a considerable hindrance that must be addressed to entirely harness its potential.
Reference:
- Okazaki Y, Fujinaga S, Salcher M M, et al. Microdiversity and phylogeographic diversification of bacterioplankton in pelagic freshwater systems revealed through long-read amplicon sequencing. Microbiome, 2021, 9: 1-15.


