
 Sample Submission Guidelines
Sample Submission GuidelinesAs a leading provider of NGS services, CD Genomics is providing an integrated portfolio of methylation sequencing services. Whole genome bisulfite sequencing (WGBS) is an effective and reliable strategy to identify individually methylated cytosines on a genome-wide scale. With over 10 years of experience and the state-of-the-art next-generation sequencing platforms, we can totally meet your project requirements and budgets in the exploration of methylome.
What Is Whole Genome Bisulfite Sequencing
In mammalian biology, 5-methylcytosine involves a covalent linkage between a methyl group and the 5' carbon position of cytosine, therein bestowing additional capacitive traits for signal transduction and regulatory functionality. As one of the predominant epigenetic modifications, 5-methylcytosine plays a pivotal role in numerous biological processes, including gene silencing, transposon repression, the inhibition of repetitive sequences, genomic imprinting, and X-chromosome inactivation. The detection and quantification of methylation are fundamentally critical for comprehending gene expression and other processes subjected to epigenetic regulation.
DNA methylation constitutes a vital part of epigenetics, playing an integral role in maintaining normal cellular functions, genetic imprinting, embryonic development, and the onset of human tumorigenesis. WGBS employing a bisulfite conversion of unmodified cytosines to uracils in genomic DNA, allows the generation of a comprehensive methylation profile. By performing a high-throughput resequencing of the converted DNA and comparing it with a reference genome, the WGBS method enables a genome-wide analysis of methylation metrics at single-base resolution, offering high precision. Its broader application spans from probing the fundamental mechanics of cellular differentiation and tissue development to advancing fields of agricultural breeding, human health, and disease treatment.
WGBS is the method of choice for drawing comprehensive maps of DNA methylation at the individual base level. This approach, often referred to as the "gold standard" for methylation sequencing, stands out due to its high throughput, time-efficiency, single-base resolution, and extensive coverage.
Principle of WGBS
The principle of WGBS relies on the treatment with heavy bisulfate to convert un-methylated cytosines in the genome into uracils, which become thymines after PCR amplification and can thus be distinguished from methylated cytosines. Coupled with high-throughput sequencing technology and referencing sequence comparison, methylation at CpG/CHG/CHH sites can be determined, making it particularly suitable for constructing single-base resolution whole-genome DNA methylation maps. For further detailed information, please refer to the article, "Principles and Workflow of Whole Genome Bisulfite Sequencing".
Advantages of WGBS
- Suitable for research on humans and most animals and plants (with known reference genomes).
- Highly integrated single-base resolution DNA methylation patterns.
- Maximally obtain comprehensive whole-genome methylation information, accurately mapping methylation patterns.
- Providing insights into gene cell fate commitment and reprogramming, as well as gene regulation.
- Identifying new epigenetic marks and disease targets.
Application of WGBS
- Gene expression regulation
- Epigenetic heterogeneity
- Environment and epigenetics
- Embryonic development
- Disease research
- Genetic imprints
- Tumor research
- Genome stability research
- Evolutionary biology research
Different DNA Methylation Detection Techniques
450K/850k
This technology scans and detects approximately 450K and 850K important CpG sites known in the genome through chip scanning. It exhibits high accuracy and repeatability, but it is limited to human samples and cannot detect new CpG sites outside of the chip on the genome.
MeDIP-seq
Methylated DNA immunoprecipitation sequencing (MeDIP-seq) enriches and sequences regions undergoing methylation, obtaining the methylation status of most methylated regions across the entire genome. It lacks single-base resolution and absolute quantification of methylation levels, only providing relative methylation levels. It is suitable for exploring the genome-wide methylation status without the need for single-base resolution.
WGBS
This technique can detect effective CpG sites reaching over 75% of all CpG sites in the entire genome, achieving single-base resolution. It is suitable for systematically and comprehensively studying the genome-wide methylation status.
RRBS
Reduced representation bisulfite sequencing (RRBS) detects the methylation status of regions rich in CG-type sites (mainly regulatory regions) in the genome, with single-base resolution and high cost-effectiveness. It is suitable for studying the methylation status of regulatory regions.
Target-BS
Target bisulfite sequencing designs primers for target fragments, performs PCR amplification after BS conversion, and finally constructs libraries for next-generation sequencing. The sequencing depth of target region sequencing can reach from hundreds to tens of thousands, enabling precise detection of methylation levels at C sites within target regions. It is used for subsequent methylation verification of target genes.
The Illumina 935K DNA Methylation Assay Service is a sophisticated tool designed for the exploration of DNA methylation. The Infinium MethylationEPIC v2.0 BeadChip (935K) facilitates high-throughput scrutiny of roughly 935,000 CpG locations dispersed throughout the human genome. This upgraded version proffers enhanced coverage and precision, ensuring congruence with the most recent genomic databases. Notable attributes of this device comprise whole-genome investigation capabilities, confirmed dependability, and broad compatibility with diverse specimen categories, encompassing formalin-fixed, paraffin-embedded tissues. The service caters specifically to comprehensive epigenetic study, providing researchers with the means to decode underlying disease mechanisms and pinpoint potential biomarkers.
The Illumina Methylation Screening Array 270K (MSA270K) represents a cutting-edge innovation tailored for meticulous methylation examinations specifically conducted within distinct disease cohorts and expansive health evaluations. Encompassing roughly 270,000 methylation loci, this tool delivers improved efficiency by allowing a throughput of 48 samples per array, constituting a sixfold increase compared to its antecedent. Empowering scientists in making strides in exploratory endeavors related to disease phenotypic patterns, environmental bearings on epigenetics, and alterations in methylation tied to aging, the MSA270K significantly bolsters our comprehension of DNA methylation and its consequential effects on human health and disease conditions. This catalyzes paradigm shifts in the fundamental understanding of our innate genetic blueprint, unlocking potential future therapeutic avenues.
WGBS Workflow
Our highly experienced expert team and strict quality control following every procedure ensure the comprehensive and accurate results. Before high-throughput sequencing, the DNA sample is processed with cytosine bisulfite conversion followed by tagging at 5' and 3' ends, and the introduction of Illumina adapters by PCR amplification.
In order to ensure the accuracy and reliability of sequencing data, CD Genomics meticulously executes each experimental step from DNA sampling to the acquisition of the final data report, through stringent quality control assessments. This comprehensive approach fundamentally guarantees high-quality data output. Securing high-quality data is the cornerstone prerequisite for accurate, comprehensive, and credible bioinformatics analysis.
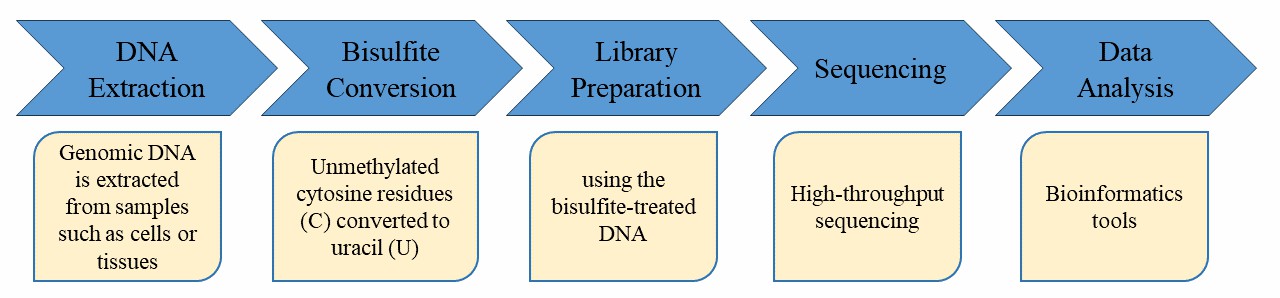
Service Specification
Sample requirements and preparation
|
|
 |
Sequencing
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
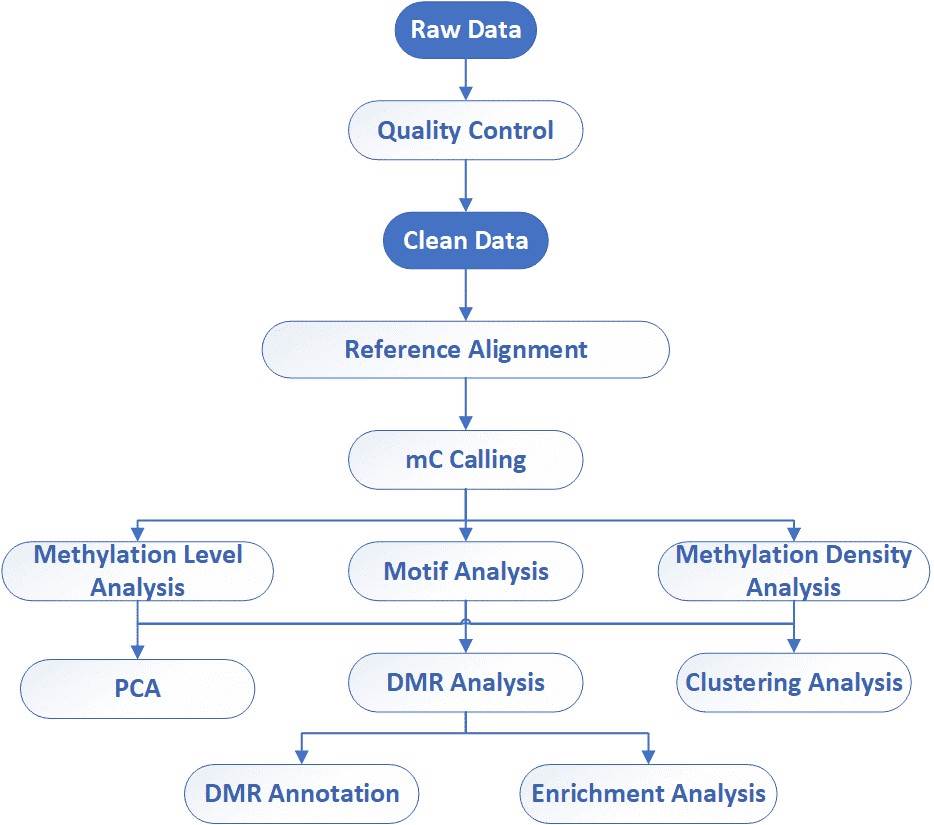
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in WGBS for your writing (customization)
Benefiting from the cutting-edge next-generation sequencing platforms and rich experience, we guarantee you high-quality data and integrated bioinformatics analyses. If you are interested in what CD Genomics can do with the whole genome bisulfite sequencing, please do not hesitate to contact us. We look forward to moving your project forward.
References:
- Huang, K., & Fan, G. DNA methylation in cell differentiation and reprogramming: an emerging systematic view. Regenerative medicine, 2010, 5(4), 531-544.
- Beck D, Ben Maamar M, Skinner M K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics, 2022, 17(5): 518-530.
- Abante J, Fang Y, Feinberg A P, et al. Detection of haplotype-dependent allele-specific DNA methylation in WGBS data. Nature communications, 2020, 11(1): 5238.
- Suzuki M, Liao W, Wos F, et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome research, 2018, 28(9): 1364-1371.
1. What species are suited for whole genome bisulfite sequencing?
The species subjected to genome bisulfite sequencing should meet the following three conditions:
a. Eukaryotes.
b. Its reference genome has been assembled to the scaffold level at least.
c. Relatively complete genome annotations.
2. What are the main contents of advanced bioinformatics analysis in WGBS?
Analysis of the correlation between methylation levels and transcriptional levels constitutes a major component of advanced bioinformatics analysis in whole-genome methylation sequencing. It primarily encompasses four aspects:
- Methylation levels of differentially expressed genes.
- Methylation levels of differentially expressed transposable elements.
- Relationship between methylation modifications and expression regulation.
- Expression analysis of methylated and non-methylated genes.
3. What are the advantages of WGBS compared to other methylation sequencing methods?
In the field of DNA methylation sequencing, current primary methodologies include Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA Immunoprecipitation (MeDIP), and Reduced Representation Bisulfite Sequencing (RRBS). WGBS boasts the advantage of allowing the evaluation of methylation status at nearly every CpG site. This assessement can extend to areas of low CpG density, including intergenic "gene deserts", partially methylated domains, and distant regulatory elements. Furthermore, WGBS can reveal absolute DNA methylation levels and the methylation sequence context. However, it should be noted that MeDIP cannot achieve single-base resolution of methylation, and RRBS typically selects areas with a higher degree of methylation for analysis, which precludes obtaining methylation status at the whole-gene level.
4. What are the limitations of WGBS?
- This method can only detect 5mC or 4mC and is unable to detect 6mA.
- Treatment with bisulfite leads to significant fragmentation of DNA, which can impact amplification.
- The conversion of unmethylated cytosines to uracils during bisulfite treatment increases the complexity of library preparation. Incomplete conversion may also introduce bias into the results.
- The method is unable to effectively distinguish between 5mC and 5hmC.
References:
- Beck D, Ben Maamar M, Skinner M K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics, 2022, 17(5): 518-530.
- Suzuki M, Liao W, Wos F, et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome research, 2018, 28(9): 1364-1371.
Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response
Journal: Scientific Reports
Impact factor: 4.259
Published: 09 October 2015
Background
DNA methylation plays a central role in the regulation of gene activity and chromatin structure in response to environmental conditions. The understanding of genome-wide DNA methylation provides insights into the regulatory mechanisms of abiotic stress response/adaptation.
Materials: Three rice (Oryza sativa) cultivars, IR64 (drought and salinity sensitive), Pokkali (salinity-tolerant) and Nagina 22 (drought-tolerant).
Methods
- Nanodrop Spectrophotometer
- Qubit Fluorimeter
- Whole genome bisulphite sequencing:
- PCR amplification
- Library quality analysis
- Primer Express (v3.0)
- Real-Time PCR System
- Gene ontology (GO) enrichment analysis
- Methylation-expression correlation analysis
Results
Researchers investigated the differences between the three cultivars in DNA methylation patterns, DMRs (Differentially methylated Regions), and gene expression, and further inquired into the underlying correlations.
1. Differential methylation is coupled to differential gene expression in different cultivars.
In comparison with all genes, those proximal to highly methylated differentially methylated regions (DMRs) demonstrate a lower level of transcript abundance, indicative of down-regulation. In contrast, genes close to lowly methylated DMRs exhibit similar or slightly higher levels of transcript abundance, suggesting an up-regulation. For DMRs, DNA methylation also appears to correlate with the on/off status of gene expression, evidenced by 14.5% in N22/IR64 and 7.2% in PK/IR64.
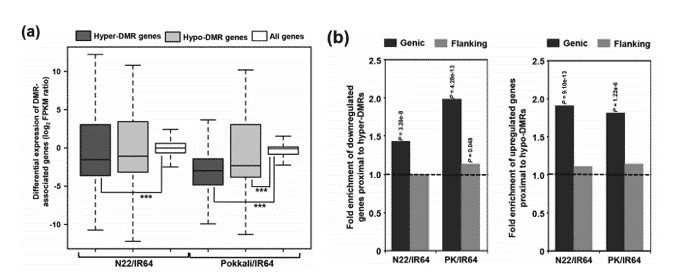 Figure 1. Relationship between differential methylation and transcript abundance of protein-coding genes.
Figure 1. Relationship between differential methylation and transcript abundance of protein-coding genes.
2. Differential methylation of genes associated with stress response and epigenetic regulation of gene expression.
The differential methylation level and corresponding differential gene expression of rice genes in N22/IR64 and PK/IR64 are shown in Fig. (a) below. The GO analysis of DMR-associated genes in both N22/IR64 and PK/IR64 revealed a significant enrichment of genes that participate in stress response.
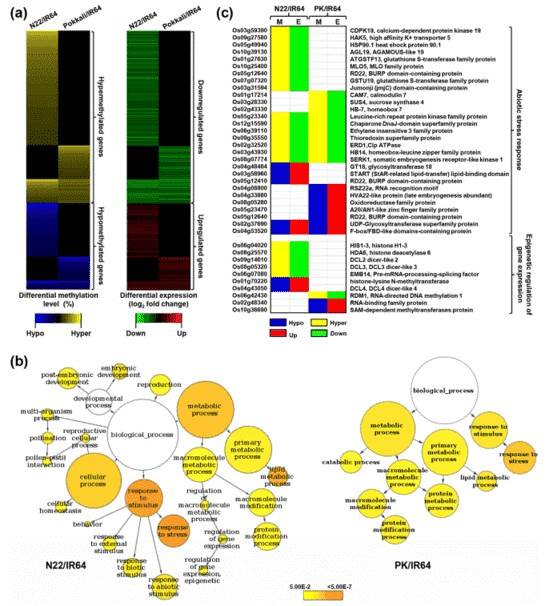 Figure 2. Differential methylation, transcript abundance and GO enrichment of proteincoding genes.
Figure 2. Differential methylation, transcript abundance and GO enrichment of proteincoding genes.
3. Methylation status of TEs is correlated with transcription of proximal protein-coding genes.
The enrichment of TEs was substantially higher for DMR-associated DEGs as compared to non-DEGs for both N22/IR64 (P-value 1.49e-05) and PK/IR64 (P-value 5.26e-05) (a). Significantly higher differences in the methylation level of DMRs are associated with TEs as compared to protein-coding genes for PK/IR64 (b).
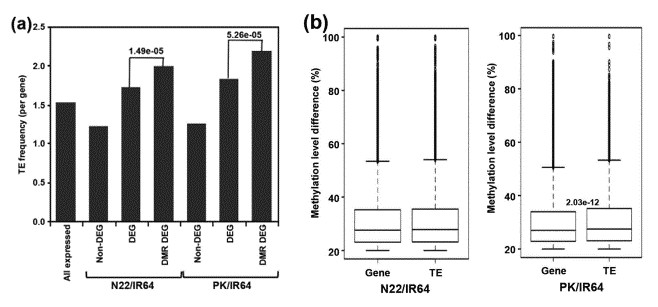 Figure 3. Correlation between differential expression and frequency/methylation level of TEs.
Figure 3. Correlation between differential expression and frequency/methylation level of TEs.
Conclusion
The results suggest the potential role of cultivar-specific DNA methylation patterns as a crucial regulatory mechanism for sensing and responding to the stress conditions via modulation of stress-responsive gene expression. The investigations suggest that variability for drought or salinity tolerance in rice germplasm rely on the extent and patterns of DNA methylation. And the evidences suggest that DNA methylation plays an important part in abiotic stress response by regulating expression of a suit of stress-responsive genes in rice mostly via methylation or demethylation of proximal TEs.
Reference:
- Garg R, Chevala V V S N, Shankar R, et al. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Scientific reports, 2015, 5.


