
 Sample Submission Guidelines
Sample Submission GuidelinesCD Genomics provides viral genome sequencing service within Illumina and PacBio Platforms. We can create high-quality de novo assembly of large viral genomes and highest possible data quality at low cost.
What Is Viral Genome Sequencing
A virus consists either of a singular nucleic acid molecule (DNA or RNA) enclosed by proteins or simply by proteins alone, as exemplified by the prion virus. The classification of viruses can be based on the composition of their genetic material, divided into single-stranded DNA viruses (ssDNA), double-stranded DNA viruses (dsDNA), single-stranded RNA viruses (ssRNA), double-stranded RNA viruses (dsRNA), and protein viruses like the prion virus. Moreover, the host type also influences the categorization, resulting in differentiations such as bacteriophages (bacterial viruses), plant viruses (tobacco mosaic virus, for instance), and animal viruses (including avian influenza virus, smallpox virus, HIV, and others).
Viral genome sequencing represents a critical technique harnessed in contemporary virology, premised on the second-generation high-throughput sequencing technologies. It involves acquiring viral genome sequences, plus interpreting the encoded information and variant data through bioinformatics analytics. A frequently utilized Next-Generation Sequencing (NGS) platform is Illumina, suggesting analysis of maximum fragment lengths approximating 300-500 nucleotides. Recently, strides were made in sequencing technology by PacBio's advent of Single-Molecule Real-Time (SMRT) sequencing. Here, complete long-reads are attainable with minimal errors, circumventing some of the limitations encountered with traditional NGS methodologies.
CD genomics platform holds great potential for viral genome sequencing. Comprehensive virus sequences will facilitate interpretation of viral metagenomics data by providing reference genomes, lead to a better understanding of virus diversity, ecology, adaptation and evolution, and enable the prediction of emerging infectious diseases caused by viruses.
Advantages of Our Viral Genome Sequencing Service
- No reference sequence required
- Sequences with considerable depth make sense for your project
- Complete characterization of viral genomes
- Identify and quantify minor variants
- Generate complete de novo assemblies of large viral genomes
- Cost-effective high-throughput sequencing
- High quality data & fast turnaround
Application of Viral Genome Sequencing
- Study of Viral Evolution and Transmission Dynamics
- Vaccine Design and Efficacy Monitoring
- Monitoring Drug Resistance
- Disease Diagnosis and Screening
- Study of Host-Virus Interactions
- Environmental Monitoring and Viral Tracin
- Genetic Engineering and Viral Therap
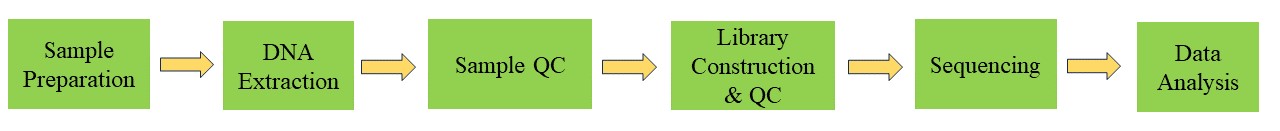
Viral Genome Sequencing Workflow
Service Specifications
Sample requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
|
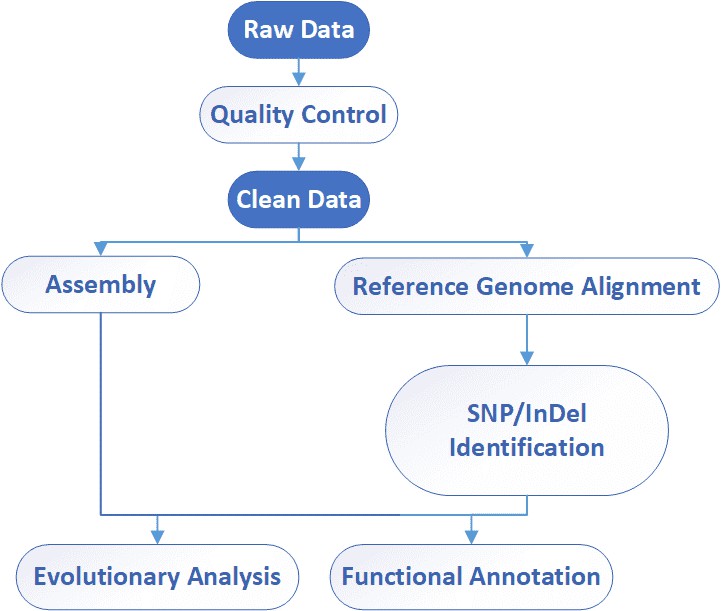
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Viral Genome Sequencing for your writing (customization)
Viral genome sequencing is a fast and efficient method for research on viral replication, packaging, function of terminase, transcription regulation, and metabolism of host cell. CD genomics can deliver high quality sequencing data for your virus genome of interest. Contact us for assistance in configuring your project.
References:
- Houldcroft C J, Beale M A, Breuer J. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology, 2017, 15(3): 183-192.
- Marston D A, McElhinney L M, Ellis R J, et al. Next generation sequencing of viral RNA genomes. BMC genomics, 2013, 14: 1-12.
1. What are the methods of viral genome sequencing?
Genomic sequencing techniques regularly utilized for the analysis of viruses encompass Whole Genome Sequencing (WGS), Targeted Sequencing, Metagenomic Sequencing, Metatranscriptomics, and Single-cell Sequencing. Each method exhibits its unique sets of advantages and disadvantages and can be appropriated selectively based on the specific research objective and empirical context.
2. How are sequence data handled during viral genome sequencing?
The sequence data generated from viral genome sequencing typically requires analysis steps including quality control, sequence alignment, variation detection, and functional annotation. Quality control processes include the removal of low-quality sequences and sequence contamination, ensuring data reliability. Subsequently, the sequence data needs to be compared with known viral genome sequences, identifying variant points within the sequence and annotating them, contributing to an understanding of potential functions and pathogenicity.
3. What is the significance of virus genome sequencing for epidemics?
Through the sequencing analysis of viral genomes, it aids us in tracing the origin and transmission pathway of viruses, pinpointing the onset of disease outbreaks, and comprehending the routes of viral dissemination and its scale. This approach bolsters the implementation of timely control measures and emergency responses, effectively curtailing the spread and proliferation of the epidemic, thus safeguarding public health and societal stability.
Consistent sequencing of viral genomes the detection of mutational changes within the virus, facilitates the study of sequence-based virus evolution, and promptly identifies emerging variant strains. This provides a preemptive warning system and offers decisive guidance for disease outbreak management. Subsequently, this valuable information forms a crucial reference for the design and development of vaccines.
Sequencing viral genomes from a single isolated plaque
Journal: Virology Journal
Impact factor: 5.913
Published: 06 June 2013
Backgrounds
The sequencing of viral and bacteriophage genomes often encounters impediments due to the substantial requirement for genomic material. The present study delineates a method that amalgamates singular plaque sequencing and sequence-independent single-primer amplification (SISPA) optimization. This methodology is applicable for next-generation whole-genome sequencing of any cultivable virus, obviating the necessity for large-scale production of viral stock or the application of centrifugation techniques for virus purification.
Methods
- Influenza virus plaque growth
- Nucleic acid extraction
- Amplification of double-stranded SISPA products
- 454 sequencing
- Illumina sequencing
- Sequence preprocessing
- Sequence assembly and mapping
- Dentification of SNPS
Results
The study's sequence data analysis shows that from an individual plaque of Influenza A, a mapping assembly with average coverage of 3590 times representing 100% of the genome can be produced. De novo assembled data create an overlap group of an average sequence coverage of 30 times, covering 96.5% of the genome. In the SISPA experimental scheme, only sequencing data using 10 pg of starting DNA from phage F_HA0480sp/Pa1651 gave a mapping assembly with average sequence coverage of 3488 times, representing 99.9% of the reference, as well as de novo assembly coverage with an average sequence coverage rate of 45 times, covering 98.1% of the genome.
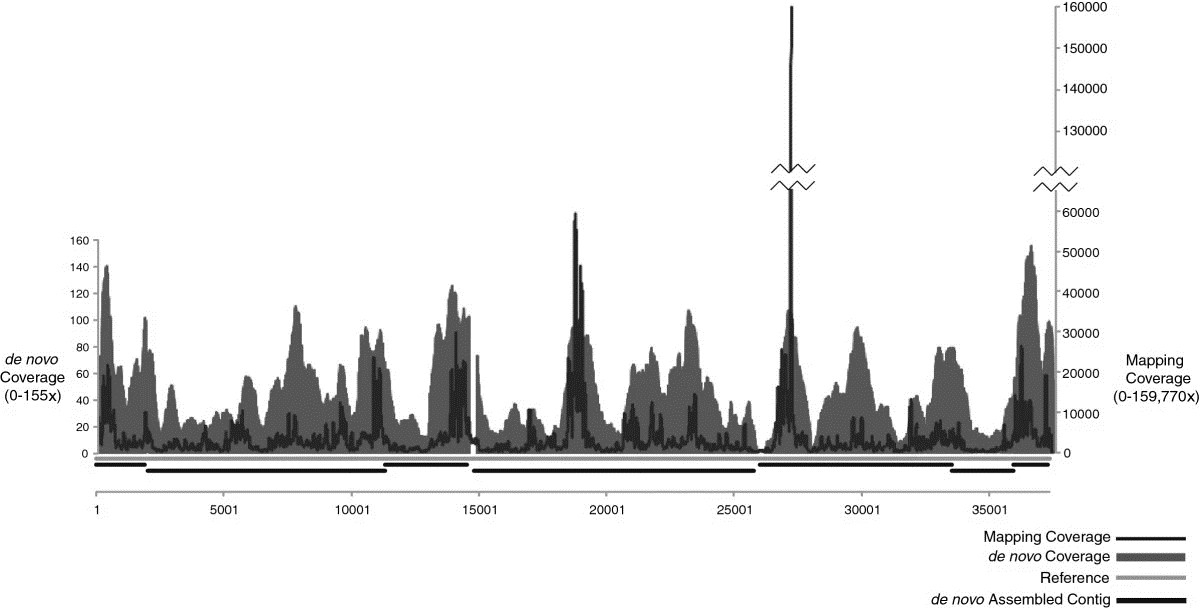 Figure1. Mapping and de novo assembly coverage sequencing results for the phage SISPA product from 10 pg of genomic DNA.
Figure1. Mapping and de novo assembly coverage sequencing results for the phage SISPA product from 10 pg of genomic DNA.
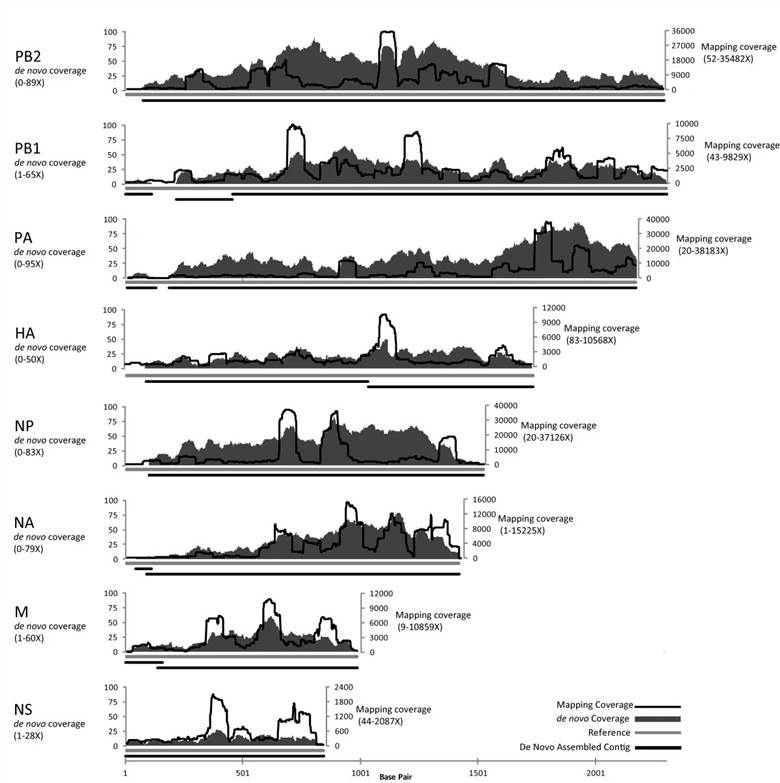 Figure 2. Mapping and de novo assembly coverage sequencing results for the flu SISPA product from a single plaque.
Figure 2. Mapping and de novo assembly coverage sequencing results for the flu SISPA product from a single plaque.
Conclusion
The paper expounds on an optimized Sequencing-Independent, Single-Primer Amplification (SISPA) protocol, capable of generating amplified products. These contribute invaluable high-quality data for de novo assembly during sequencing. The approach requires merely a single viral plaque or a minimal template of 10 pg of DNA, facilitating the prompt identification of viruses during outbreaks and those with limited transmission. This aspect underscores the potential of the method in navigating challenges in virus detection and control.
Reference:
- DePew J, Zhou B, McCorrison J M, et al. Sequencing viral genomes from a single isolated plaque. Virology journal, 2013, 10: 1-7.


